
Lékové interakce a současná klinická praxe
Drug interaction and current clinical practice
Drug interaction is defined as an elevation, lowering or other influencing of the effect of one drug by another drug or substance. The aim of the article is to describe undesired drug interactions that are a source of adverse effects, sometimes even life ‑ threatening. The incidence of important drug interactions varies between 4% and 88%. The risk of drug interactions increases with the number of used drugs. According to the causing mechanism, drug interactions are divided into pharmaceutic, pharmacokinetic and pharmacodynamic, however, particular drug interactions can mutually overlap and combine. Pharmacokinetic drug interactions occur at the level of absorption, distribution and elimination that consists of biotransformation and excretion. At the level of biotransformation, induction or inhibition of enzymes of cytochrome P450 is very common. Most common inductors include rifampicin, most antiepileptic drugs, barbiturates, St John’s wort and smoking of cigarettes. Inhibitors include calcium channel blockers diltiazem and verapamil, amiodarone, most macrolide antibiotics and most azole antifungal drugs and grapefruit juice. The metabolic activity of enzymes of cytochrome P450 is in some cases influenced by polymorphism of the genes which code them. The most important polymorphic genes of cytochrome P450 which are routinely investigated are the genes of enzymes CYP2C9, CYP2C19 and CYP2D6. Clinically significant drug interactions are most often caused by warfarin. Since the prevalence of drug interactions will probably rise in future, it is necessary to increase the knowledge of the topic among professionals as well as the non‑professional public.
Keywords:
drug interactions – pharmacokinetics – pharmacodynamics – cytochrome P450 – pharmacogenetics – inhibition – induction
Autoři:
J. Holý; M. Pešková
Působiště autorů:
Interní oddělení, Nemocnice České Budějovice, a. s.
Vyšlo v časopise:
Kardiol Rev Int Med 2015, 17(1): 70-75
Kategorie:
Interní medicína
Souhrn
Léková interakce je definována jako zvýšení, snížení či jiné ovlivnění účinku jednoho léčiva podáním jiného léčiva či látky. Cílem článku je popsat lékové interakce nežádoucí, které jsou zdrojem nežádoucích účinků, někdy i život ohrožujících. Incidence významných lékových interakcí kolísá v rozmezí 4 – 88 %. Riziko lékových interakcí stoupá s počtem užívaných léků. Lékové interakce se podle vyvolávajícího mechanizmu dělí na farmaceutické, farmakokinetické a farmakodynamické, jednotlivé lékové interakce se však mohou vzájemně překrývat i kombinovat. K farmakokinetickým lékovým interakcím dochází na úrovni absorpce, distribuce a eliminace, která se skládá z biotransformace a exkrece. Na úrovni biotransformace dochází často k indukci nebo inhibici enzymů cytochromu P450. K induktorům patří rifampicin, většina antiepileptik, barbituráty, třezalka tečkovaná a kouření cigaret. K inhibitorům patří z blokátorů kalciových kanálů diltiazem a verapamil, dále amiodaron, většina makrolidových antibiotik a azolových antimykotik a grapefruitový džus. Metabolická aktivita enzymů cytochromu P450 je v některých případech ovlivněna polymorfizmem genů, které je kódují. Nejdůležitějšími polymorfními geny cytochromu P450, které se rutinně vyšetřují, jsou geny enzymů CYP2C9, CYP2C19 a CYP2D6. Nejčastěji vyvolává klinicky závažné lékové interakce warfarin. Jelikož bude prevalence lékových interakcí v budoucnu zřejmě stoupat, je nutné zvyšovat informovanost odborné i laické veřejnosti o tomto tématu.
Klíčová slova:
lékové interakce – farmakokinetika – farmakodynamika – cytochrom P450 – farmakogenetika – inhibice – indukce
Úvod
Lékovou interakcí (LI) nazýváme zvýšení, snížení či jiné ovlivnění účinku jednoho léčiva podáním jiného léčiva či látky. Podání tohoto jiného léčiva či látky může podání léčiva předcházet, může proběhnout současně i následně [1]. Změny v síle a trvání účinku léčiva vlivem LI jsou měřitelné. Léčivo vyvolávající LI může být léčivým přípravkem jak vydávaným na předpis, tak volně prodejným. Látkou vyvolávající LI může být alkohol a součásti potravy, například mléko a mléčné produkty, nápoje obsahující kofein a flavonoidy v grapefruitovém džusu [2], může však jít i o chemickou látku zevního prostředí [3]. LI jsou v určitých případech prospěšné a využívají se. Příkladem takového synergického účinku lékových kombinací může být léčba protinádorová, imunosupresivní, antihypertenzní, antiastmatická i léčba infekčních onemocnění [1]. Cílem tohoto článku je však popis LI, které mohou být zdrojem nežádoucích účinků, a to i život ohrožujících. Příkladem může být LI hypnotika ramelteon s antidepresivem fluvoxamin. Pokud si nemocný užívající antidepresivum fluvoxamin v dávce 2 tablety denně po dobu tří dnů vezme 1 tabletu hypnotika ramelteon, dojde k rozšíření plochy pod křivkou ramelteonu (AUC – aera under curve) 190× a zvýšení jeho maximální koncentrace (Cmax) 70× [4]! Takový nemocný se tedy ocitne v situaci jedince, který byl intoxikován 190 tabletami výše zmíněného hypnotika! Tento příklad ilustruje jak závažnost, tak aktuálnost tématu LI. LI mohou být též příčinou stažení léčivého přípravku z trhu. Z důvodů LI, které měly v některých případech fatální důsledky, byl v roce 1998 stažen z trhu blokátor kalciového kanálu mibefranil [5]. Z důvodu rabdomyolýzy, která vznikla v důsledku léčby hypolipidemikem cerivastatin často v kombinaci s fibrátem gemfibrozil, bylo toto hypolipidemikum v roce 2001 též staženo z trhu [6]. Nelze nezmínit, že téma LI patří v tuzemsku k tématům tradičním, a to již od poloviny 70. let minulého století [7].
Dělení a význam lékových interakcí
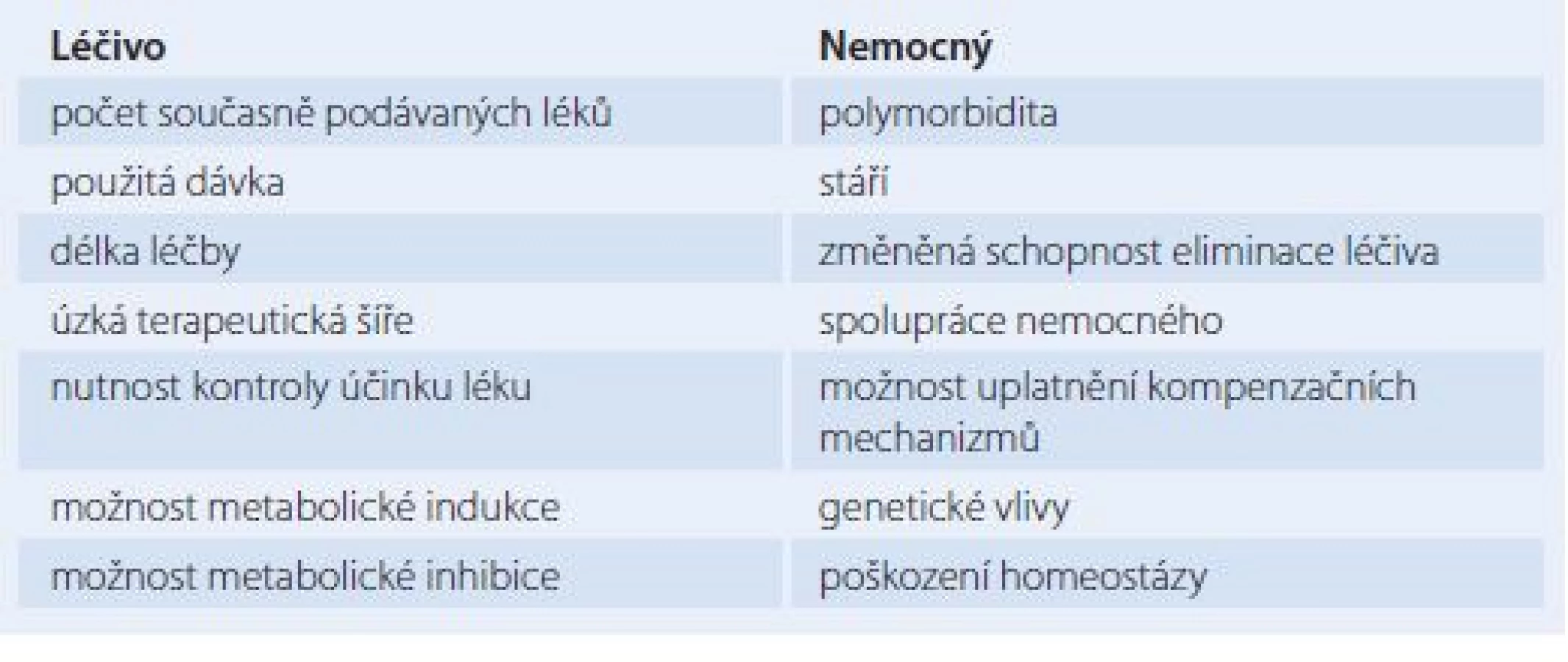
Lékové interakce se podle vyvolávajícího mechanizmu dělí na farmaceutické, farmakokinetické a farmakodynamické. Při farmaceutických LI dochází ke vzájemnému fyzikálně chemickému či chemickému ovlivnění buď ještě mimo organizmus, či na úrovni vstupní cesty do organizmu. Při farmakokinetických LI dochází k vzájemnému ovlivnění léčivých látek v organizmu při cestě k cíli jejich účinku, k orgánům eliminace a při vlastní eliminaci z těla. Lze tedy rozlišit LI na úrovni absorpce, biodistribuce a eliminace, která se skládá z biotransformace a exkrece. Na úrovni biotransformace často dochází k enzymové indukci nebo enzymové blokádě čili inhibici. Pod pojmem enzymová indukce rozumíme zvýšení syntézy a aktivity enzymu, který se zúčastňuje biotransformace léčiv, a to vlivem opakovaného podávání určitého léčiva – induktoru. Tento jev se týká především oxidáz dependentních na cytochromu P450. Pokud induktor ovlivní svou vlastní biotransformaci, mluvíme o autoindukci, pokud ovlivní biotransformaci jiných léčiv, jde o heteroindukci. Důsledkem indukce je snižování intenzity a zkracování doby účinku léčiva, které je enzymem biotransformováno. Opačným jevem je enzymová inhibice, event. i blokáda v důsledku opakovaného podávání léčiv. Enzymová inhibice vede k zesílení účinku léčiv až k intoxikaci [2]. Mechanizmus enzymové inhibice spočívá v kompetici mezi substráty či v ireverzibilní inaktivaci enzymu vlivem inhibitoru [8]. Při farmakodynamických LI dochází ke změnám účinku na úrovni receptorů. Farmakodynamické LI jsou tedy způsobeny kompeticí dvou látek na jednom receptoru nebo společným působením na stejný fyziologický systém [1,2]. Typy LI vyjmenované výše se však mohou vzájemně překrývat i kombinovat. Jinak lze také LI klasifikovat podle konečného léčebného dopadu, kterým může být antagonizmus, synergizmus, ovlivnění vedlejších účinků některého z interagujících látek či vznik vedlejšího účinku nového [8]. Odhad prevalence LI v praxi je velice obtížný, neboť jsou těžko rozpoznatelné a neevidují se. Některé LI se mohou snadno zaměnit za komplikace, které vyplývají z průběhu léčeného patofyziologického stavu. Dle farmakoepidemiologických studií je incidence prokazatelných následků LI uváděna v širokém rozpětí 4 – 88 % [9]. U hospitalizovaných nemocných byl dle analýzy preskripce výskyt klinicky významných LI odhadnut na 4 – 5 %, dle další studie byla LI bezprostřední příčinou hospitalizace u téměř 1 % [3]. Dle analýzy údajů u 400 000 ambulantních nemocných se příznaky vyplývající pravděpodobně z LI objevily u 11,1 % [3]. Faktory, které zvyšují pravděpodobnost výskytu LI, lze rozdělit na faktory týkající se léčiva a na faktory týkající se nemocného. Tyto faktory uvádí tab. 1. Je prokázáno, že výskyt LI stoupá s počtem současně podávaných léků, a to prakticky exponenciálně. Pokud užívá nemocný kombinaci do pěti léků, je výskyt nežádoucích LI 4 %, pokud jich však užívá 16 – 20, je výskyt 54 % [9]. Problematika LI je složitá též kvůli jejich interindividuální variabilitě. Tato variabilita znamená, že se LI nemusí klinicky projevit u všech nemocných [3]. Zjistíme‑li možnost potenciálně klinicky významné LI určité komedikace, je v určitých případech nutno jeden z léků nepodávat vůbec či jej vysadit. V jiných případech je indikována úprava dávky, a to zejména při zahájení či při ukončení terapie léčivem vstupujícím do LI. Jindy stav nemocného klinicky, popř. laboratorně monitorujeme. V tomto případě představuje účinnou pomoc tzv. terapeutické monitorování léčiv (TDM – therapeutic drug monitoring) [3]. Cílem TDM je optimalizace dávkování léku na základě jeho změřené koncentrace, zvážení klinického stavu nemocného s přihlédnutím k farmakokinetice léčiva a k současně podávaným lékům. TDM výrazně snižuje nebezpečí jak předávkování, tak poddávkování lékem [10]. Základním zdrojem informací o LI konkrétního léčivého přípravku je souhrn údajů o přípravku (SPC – summary of product characteristics), který lze nalézt na internetových stránkách registračních orgánů. Existuje několik internetových zdrojů informací o inhibitorech, induktorech a substrátech na úrovni cytochromu P450. Cenným praktickým tuzemským pomocníkem v problematice LI je pravidelně aktualizovaný program společnosti Infopharm, a. s., Praha. Pomocí tohoto programu lze jednak zjistit potenciální známé LI konkrétní kombinace léčiv, jednak obsahuje edukační materiál. Program celkově hodnotí každou LI ve škále 0 – 5, přičemž při hodnocení 0 k LI prokazatelně nedochází, při hodnocení 5 jde o tak významnou LI, že představuje kontraindikaci takovéto testované komedikace [11].
Farmaceutické lékové interakce
V případě farmaceutických LI dochází k vzájemnému ovlivnění na fyzikálně chemické či chemické bázi mimo organizmus (tedy v infuzní lahvi či injekční stříkačce) či na úrovni vstupní cesty do organizmu ještě před absorpcí. Farmaceutické LI jsou tedy vlastně inkompatibilitami. Prevencí vzniku inkompatibilit se zabývají již při výrobě hromadně vyráběných léčivých přípravků farmaceutičtí technologové. Informace o případných inkompatibilitách již vyrobeného léčiva lze zjistit v příslušném SPC. Problematika inkompatibilit musí být známa i lékárníkovi, který zhotovuje a vydává individuálně připravované léčivé přípravky [2]. Kromě SPC a tištěné odborné literatury je při orientaci v této problematice také pomocníkem internet [12]. Co se týče inkompatibilit, ke kterým dochází ještě mimo organizmus a které jsou aktuální vzhledem k současné klinické praxi, je třeba zmínit LI mezi betalaktamovými antibiotiky (ATB) a aminoglykosidy. Tato ATB se nesmějí mísit v jedné stříkačce či infuzní lahvi, ovšem vzhledem k možnosti LI i při mísení v krvi je třeba je podávat po sobě vždy s určitým časovým odstupem. V opačném případě totiž hrozí riziko snížení účinku. Riziko precipitace hrozí též při mísení vankomycinu s betalaktamovými ATB v jedné stříkačce či infuzní lahvi. Tato kombinace ATB by též neměla být podávána intravenózně hned po sobě, ale minimálně po proplachu infuzního setu [13]. Vzhledem k riziku precipitace nelze též v jedné infuzní lahvi před intravenózním podáním mísit ketoprofen a tramadol [14]. Co se týče aktuálních farmaceutických LI, ke kterým dochází již v organizmu, ale ještě před absorpcí, je potřeba zmínit zejména reakci perorálně podávaných tetracyklinů (deoxymykoinu) s ionty hořčíku, vápníku a hliníku obsaženými v antacidních směsích, při které dochází ke vzniku nevstřebatelných komplexů [2,9]. Při užívání deoxymykoinu je ze stejného důvodu potřeba se vyvarovat také potravin s obsahem vápníku. S deoxymykoinem mohou interagovat i přípravky obsahující železo, aktivní uhlí a cholestyramin [15]. Ionty obsažené v antacidních směsích i ionty železa obsažené v antianemikách tvoří s ciprofloxacinem cheláty, které nemohou být vstřebány [9].
Farmakokinetické lékové interakce na úrovni absorpce z trávicího traktu
K LI na úrovní absorpce z trávicího traktu dochází pochopitelně jen při perorálním podání léčiva, které je tou nejfyziologičtější cestou jeho přívodu do organizmu. Absorpce léčiva se děje pasivní difuzí lipoproteinovou membránou či aktivním transportem. Absorpce závisí na charakteru lékové formy, na liposolubilitě léčiva, na pH v jednotlivých oddílech trávicího traktu, na jeho motilitě a biotransformační kapacitě, dále na bakteriální flóře ve střevě a na průtoku krve zásobující střevo [9]. LI na úrovni absorpce se mohou týkat jak rychlosti, tak rozsahu vstřebávání léčiva. Změna pH se projeví zejména na absorpci slabých kyselin, které se vstřebávají již v žaludku: tak látky zvyšující žaludeční pH (blokátory protonové pumpy, H2 blokátory, antacida) snižují absorpci např. kyseliny acetylosalicylové [9]. Omeprazol snižuje vstřebávání perorálně podaných azolových antimykotik (flukonazolu, itrakonazolu a posakonazolu) [16]. Absorpci některých léčiv ovlivňuje současný příjem stravy či nápojů. Pryskyřice typu cholestyraminu, kaolin či carbo medicinalis mohou brzdit absorpci látek, které se na ně v proximální části trávicího traktu naváží. Týká se to např. digoxinu, warfarinu, cyklosporinu A či tricyklických antidepresiv [9]. Z obdobného důvodu je potřeba antidiarhoikum diosmectic (Smecta) podávat s časovým odstupem od ostatních léků [17]. Látky ovlivňující motilitu trávicího traktu mají vliv především na rychlost absorpce. Metoklopramid urychluje motilitu žaludku, a tak zvyšuje rychlost absorpce i Cmax těch léčiv, která se vstřebávají ve střevě, např. cyklosporinu A, paracetamolu či diazepamu. Absorpce digoxinu je však vlivem metoklopramidu naopak snížena. Zpomalení motility trávicího traktu anticholinergiky (např. typu propanthelinu) sníží rychlost absorpce levodopy, paracetamolu a diazepamu [3]. Pasáž trávicím traktem však zpomalují i tricyklická antidepresiva, fenotiaziny či některá antihistaminika [9]. Malabsorpční syndrom vyvolaný léky (neomycin kolchicin, cytostatika) zapříčiňuje snížené vstřebávání digoxinu, V ‑ penicilinu a vitaminu B12. Cisplatina a anticyklinová antibiotika snižují absorpci např. fenytoinu [9,3]. Po podání širokospektrých antibiotik dochází k ovlivnění farmakokinetiky těch léčiv, která jsou biotransformována střevní flórou, např. sulfasalazinu a digoxinu [9].
Farmakokinetické lékové interakce na úrovni biodistribuce
K LI na úrovni biodistribuce dochází zejména na úrovni vazby na plazmatické bílkoviny. Léčivo se v krvi nachází jednak ve frakci volné, která je účinná, jednak ve frakci vázané, která se reverzibilně váže na plazmatické proteiny či na krevní elementy a která vytváří „přechodné depo“. Pokud bylo dosaženo ustáleného stavu, nastává mezi těmito frakcemi rovnovážný stav. Na plazmatické albuminy se váží především slabé kyseliny (např. kyselina acetylosalicylová, nesteroidní antiflogistika, warfarin, diazepam a jiné benzodiazepiny), na kyselé alfa 1 glykoproteiny slabé baze (např. tricyklická antidepresiva). Podle warfarinu a diazepamu dostala vazebná místa na albuminu svá jména, na tato místa se však váží i jiná léčiva, z nichž některá dokonce na obě (např. acidum salicylicum) [9,18]. Jelikož se některá léčiva váží na stejná vazebná místa, mohou se z vazby vzájemně vytěsňovat. Léčivo z vazby na bílkoviny vytěsněné zvýší svoji volnou, účinnou frakci. Klinicky významné zvýšení účinku však nastává pouze u léčiv s malým distribučním objemem a s výraznou vazbou na bílkoviny převyšující 90 %. Zvýšení účinku léčiva je navíc přechodné, neboť volná část léčiva se z organizmu současně snáze eliminuje, v některých případech může však jít i o efekt toxický [1]. K léčivům s významnou vazbou na plazmatické bílkoviny patří warfarin a fenytoin [3]. Dle SPC kolísá plazmatická vazba warfarinu na sérový albumin od 99,5 % k 97 %. Sníží‑li se tato plazmatická vazba warfarinu z 99 % na 95 %, zvýší se jeho účinek čtyřnásobně! K závažným následkům však dojde spíše v případě postižení metabolizmu v játrech či postižení renální exkrece [9,18]. K vytěsnění z vazby však nemusí docházet jen v plazmě, ale i ve tkáních. Amiodaron spolu s verapamilem a nifedipinem zvyšují plazmatickou hladinu digoxinu jednak snížením jeho eliminace, jednak jeho vytěsněním z vazebných míst v srdečním a kosterním svalu [9]. Amiodaron tak zvýší hladinu digoxinu o 75 – 104 %, u některých nemocných však dokonce 3 – 4násobně [18].
Farmakokinetické interakce na úrovni biotransformace
Nejdůležitějším orgánem biotransformace jsou játra a u některých léčiv ledviny, méně často se na metabolizmu léčiv podílí další tkáně, např. plíce, plazma a střevní stěna. Při biotransformaci dochází jak k biodegradaci léčiv, kdy vznikají metabolity více hydrofilní, a tedy snáze vylučitelné z organizmu, tak k bioaktivaci, při které se proléčivo stává účinnou látkou. Některá léčiva se však vylučují metabolicky nezměněná [2]. Z hlediska LI na úrovni biotransformace jsou nejvýznamnější a nejprostudovanější děje katalyzované enzymy dependentními na cytochromu P450 (standardně uváděné pod zkratkou CYP), které také vykazují největší interindividuální variabilitu [9,19]. Tyto enzymy se nalézají především v játrech, dále v tenkém střevě, mozku, plicích a ledvinách. V buňce se CYP nacházejí v endoplazmatickém retikulu a v mitochondriích. Označení konkrétního cytochromu z rodiny P450 se skládá z předpony CYP, pak následuje arabská číslice označující rodinu, dále velké písmeno označující podrodinu a další arabská číslice pro označení konkrétního enzymu. Dosud je známo 14 rodin, z nichž nejdůležitější pro metabolizmus xenobiotik mají první tři rodiny [9]. Enzym CYP3A4 se podílí na biotransformaci nejvíce, a to z 53 % [9]. Substráty CYP3A4 se překrývají se substráty P ‑ glykoproteinu, který patří k proteinovým přenašečům zodpovědným za mnohočetnou lékovou rezistenci (multi‑drug resistence – MDR) [19 – 21]. U některých enzymů CYP byl prokázán polymorfizmus. Jde o přítomnost několika typů alel pro daný enzym, které předurčují různou aktivitu kódovaného enzymu. U nositelů jednoho typu alely se tvoří enzym s rychlým metabolizmem substrátů, jde o tzv. rychlé metabolizátory, nositelé alely způsobující značné snížení metabolické aktivity jsou pomalí metabolizátoři. Enzym je polymorfní, pokud se v populaci vyskytuje více než 1 % pomalých metabolizátorů [19,20]. Polymorfizmus je dobře popsán u genů kódujících CYP2A6, CYP2C9, CYP2C19, CYP2D6 a CYP2E1 [9,20]. Pracoviště molekulární genetiky v rámci farmakogenetického vyšetření většinou již rutinně stanovují geny pro CYP2C9, CYP2C19 a CYP2D6, které patří k nejdůležitějším polymorfním genům [19]. Klinický význam polymorfizmu může být značný. Variantní alela v genu CYP2C9, díky které se tvoří enzym metabolizující léčiva pomalu, může při léčbě hypoglykemizujícími antidiabetiky zapříčinit závažné hypoglykemie [20]. Tento efekt byl popsán u glipizidu [19]. U pomalých metabolizátorů omeprazolu, který je metabolizován enzymem CYP2C19, byly pozorovány 12× větší hodnoty AUC tohoto léčiva než u rychlých metabolizátorů [20]. Pomalých metabolizátorů CYP2C19 je v euroatlantické populaci do 3,8 % [22]. Další problém vyplývá z toho, že u enzymů CYP dochází často k enzymové indukci nebo inhibici. K nejdůležitějším enzymatickým induktorům patří rifampicin, většina antiepileptik a barbituráty (dnes užívané prakticky výhradně v epileptologii), z rostlinných léčiv třezalka tečkovaná (hypericum peroforatum), z chemických látek zevního prostředí pak cigaretový kouř [1,3]. Jako příklad zrychleného odbourávání léčiva vlivem indukce lze uvést selhání imunosupresivní léčby cyklosporinem A a takrolimem po podání třezalkového extraktu či selhání perorální antikoncepce s etinylestradiolem při léčbě rifampicinem či barbituráty [9,21,23]. K nejvýznamnějším enzymatickým inhibitorům patří blokátory kalciových kanálů (např. diltiazem a verapamil), amiodaron, makrolidová antibiotika (erytromycin a klaritromycin), azolová antimykotika (itrakonazol, vorikonazol) a grapefruitový džus [9,21]. Příkladem je inhibice CYP3A4 klaritromycinem, kvůli které dochází ke zvýšení účinku warfarinu, a tím k riziku krvácení [24]. Warfarin je tvořen racemickou směsí dvou stereoizomerů, z nichž je méně účinný R ‑ warfarin metabolizován CYP3A4 a CYP1A2, S ‑ warfarin pak CYPC29 [25,26]. Snížení metabolizmu S ‑ warfarinu díky inhibici enzymu CYP2C9 je klinicky významnější, pokud nastane u pomalého metabolizátora [27]. Warfarin je léčivem, které vyvolává klinicky závažné LI nejčastěji, a to i život ohrožující [28]. Důležité substráty, inhibitory a induktory jednotlivých enzymů CYP uvádí tab. 2. Závažná enzymová inhibice však může nastat i u jiných enzymů než CYP. Kvůli inhibici xanthinoxidázy alopurinolem dochází ke zvýšení koncentrací účinných metabolitů azathioprinu, což vede ke zvýšení rizika myelotoxicity [2].

Farmakokinetické interakce na úrovni biliární exkrece
Vylučovaná látka prochází při exkreci játry krevním, a poté žlučovým pólem hepatocytu. Tak jsou vylučovány především metabolity léčiv. Transport hepatocytem se děje jednak pasivní difuzí, dále též aktivním transportem, kterým jsou vylučovány větší molekuly látek rozpustných ve vodě. Po jejich transportu do střeva může dojít k jejich hydrolýze se vznikem lipofilní molekuly, která se vstřebává do portálního krevního oběhu a podléhá enterohepatální cirkulaci [2]. LI na úrovni biliární exkrece nepatří k častým LI. Klinicky závažnou LI je kompetice paclitaxelu s doxorubicinem o stejný biliární exkreční mechanizmus, jehož důsledkem je snížení clearance doxorubicinu o 30 % a zvýšení incidence jeho nežádoucích účinků. Je‑li kombinace paclitaxelu a antracyklinu indikována, lze zvýšení toxicity antracyklinu předejít intravenózním podáním antracyklinu před podáním paclitaxelu [29]. Na úrovni biliární exkrece snižuje urikosurikum probenecid clearance tuberkulostatika rifampicinu. Verapamil, amiodaron a chinidin snižují biliární clearance dioxinu, a prodlužují tak jeho účinek [3].
Farmakokinetické interakce na úrovni renální exkrece
Léčivo se v ledvinách vylučuje glomerulární filtrací a tubulární sekrecí. Glomerulární filtrace léčiva přes glomerulární kapiláry je možná jen u léčiva ve volné frakci. Logicky tedy každá LI na úrovni biodistribuce v krvi ovlivní i míru glomerulární filtrace. Další možností ovlivnění glomerulární filtrace je lékově navozené snížení či zvýšení perfuzních poměrů ve vas afferens [9]. Tubulární sekrece se děje aktivním transportem. Tímto mechanizmem jsou vylučovány jak organické kyseliny (furosemid, thiazidy, metotrexát a peniciliny), tak organické baze (např. morfin a amilorid). Na této úrovni může docházet ke kompetici látek o transportní přenašeče. Kvůli kompetici thiazidů a kyseliny močové na této úrovni může dojít u nemocných se dnou k záchvatu [2]. Kvůli kompetici o aktivní transportní, „carrierový“ systém zvyšují nesteroidní antiflogistika toxicitu metotrexátu a amiodaron s verapamilem snižují renální clearance digoxinu [3,9]. Dále mohou léčiva v ledvinách podléhat tubulární reabsorpci. Tento pasivní děj se týká látek liposolubilních (např. diazepamu), které jsou kvůli tomuto ději vylučovány velmi pomalu. Dále je pro tubulární reabsorpci důležité, že rozpustnost slabých zásad a slabých kyselin v tucích je závislá na stupni ionizace, a tedy na pH prostředí [2]. Je‑li léčivo charakteru slabé kyseliny (např. barbiturát) v kyselé moči, vzroste podíl jeho neionizované složky a dojde k jeho reabsorpci. Naopak, v alkalické moči bude tato látka ionizována více a rychleji se močí vyloučí. Léčiva charakteru slabé báze se budou chovat obráceně. Farmakokinetika těchto léčiv může být tedy ovlivněna jak okamžitou dietou, tak léčivy měnícími pH moči, např. C ‑ vitaminovou terapií [9]. Jinou situací je lékové poškození renálních funkcí vedoucí ke zhoršení vylučování dalších léčiv: aminoglykosidy tak mohou snížit exkreci digoxinu a cyklosporin A exkreci aminoglykosidů [9].
Farmakodynamické lékové interakce
Při farmakodynamických LI dochází ke změnám účinku na úrovni receptorů nebo společným působením na stejný fyziologický systém [1,2]. Farmakodynamické účinky jsou častější než farmakokinetické, ovšem na základě znalostí farmakologie jsou obvykle lépe předvídatelné [1]. Zřejmě nejzávažnějším problémem je současné podávání látek, které tlumí centrální nervový systém. Tato LI je zvláště významná u seniorů. Příkladem může být současné podání alkoholu s benzodiazepiny nebo jinými léčivy způsobujícími sedaci [3]. Tato LI je příkladem synergizmu. Antagonizmus nastává např. při komedikaci betablokátoru s verapamilem či diltiazemem, důsledkem takovéto komedikace je bradykardie, AV blokáda či srdeční selhání. Nejrizikovější je pak podání této kombinace intravenózně [2,3]. Účinek antidiabetik je snižován thiazidy, glukokortikoidy i perorálními kontraceptivy [3]. Jinou farmakodynamickou LI je zvýšení toxicity digoxinu při hypokalemii způsobené kličkovým či thiazidovým diuretikem. Jde o LI na úrovni vazby na Na+K+ ATPázu, na kterou se při hypokalemii váže zvýšenou měrou digoxin [2]. Pravděpodobně nejrozšířenější LI je oslabení účinku antihypertenziv (zejména betablokátorů, diuretik a inhibitorů angiotenzin‑konvertujícího enzymu) vlivem nesteroidních protizánětlivých léčiv, a to zřejmě díky inhibici biosyntézy prostaglandinů. Prostaglandiny totiž působí vazodilatačně na cévy v ledvinách [2,30]. Příklady potenciálně fatálních LI uvádí tab. 3.

Závěr
Z výše uvedeného přehledu vyplývá, že téma nežádoucích LI představuje významný a složitý problém. LI mohou být příčinou hospitalizace, mohou ji prodloužit a mohou mít i fatální důsledky. Problematiku LI komplikuje i jejich interindividuální variabilita a jejich obtížné klinické rozpoznání. Lze předpokládat, že s prodlužující se střední délkou života se bude zvyšovat i počet léčiv užívaných nemocnými, a tak stoupne i prevalence výskytu LI. Význam edukace odborné veřejnosti ohledně LI tedy roste. Ovšem vzhledem k tomu, že s léčivy předepisovanými lékařem interagují i léky volně prodejné, složky potravy, nápoje i chemické látky zevního prostředí, je neméně důležitá i edukace laické veřejnosti.
Doručeno do redakce: 16. 1. 2015
Přijato po recenzi: 2. 2. 2015
MU Dr. Jiří Holý, Ph.D.
www.nemcb.cz
holy@nemcb.cz
Zdroje
1. Perlík F. Klinická farmakologie v praxi. 1st ed. Praha: Triton 1999.
2. Martínková J, Chládek J, Mičuda S et al. Obecná farmakologie jako základ studia farmakologie experimentální a klinické. 1st ed. Hradec Králové: Olga Čermánková 2001.
3. Perlík F, Martínková J. Lékové interakce. Postgraduální medicína 2002; 4 : 311 – 317.
4. Souhrn údajů o přípravku Fevarin 100. [online] Dostupné z: http:/ / www.sukl.cz/ modules/ medication.
5. Stolberg SG. Heart drug withdrawn as evidence shows it could be lethal. [online] Available from: http:/ / www.nytimes.com.
6. Furberg CD, Pitt B. Withdrawal of cerivastatin from the world market. Curr Control Trials Cardiovasc Med 2001; 2 : 205 – 207.
7. Květina J, Fendrych Z. Farmakologické interakce. Praha: Avicenum 1978.
8. Katzung GB. Základní a klinická farmakologie. 2nd ed.Jinočany: H & H Vyšehradská, s. r. o. 2006.
9. Květina J, Grundmann M. Farmakologické interakce. Klin Farmakol Farm 2003; 1 : 17 – 21.
10. Šedivý J. Terapeutické monitorování hladin léčiv. Postgraduální medicína 2002; 4 : 318 – 323.
11. Suchopár J, Prokeš M, Oppeltová M et al. (Infopharm a.s. Praha). Kontrolní modul lékových interakcí INFOPHARM. [online] Dostupné z: http:/ / www.drugagency.cz.
12. Vigneron J, Gindre I, Daouphars M et al. Stabilis. [online] Available from: http:/ / www.stabilis.org.
13. Souhrn údajů o přípravku Vancomycin Hikma 500 a 1000 mg prášek pro koncentrát pro infuzní roztok. [online]. Dostupné z: http:/ / www.sukl.cz/ modules/ medication.
14. Souhrn údajů o přípravku Ketonal injekční roztok. [online] Dostupné z: http:/ / www.sukl.cz/ modules/ medication.
15. Souhrn údajů o přípravku Doxybene 100 mg měkké tobolky. [online] Dostupné z: http:/ / www.sukl.cz/ modules/ medication.
16. Souhrn údajů o přípravku Helicid 20 Enterosolventní tvrdé tobolky. [online] Dostupné z: http:/ / www.sukl.cz/ modules/ medication.
17. Souhrn údajů o přípravku SMECTA prášek pro perorální suspenzi. [online] Dostupné z: http:/ / www.sukl.cz/ modules/ medication.
18. Grundmann M. Lékové interakce II. Int Med Prax 2000; 2 : 39 – 40.
19. Rogers JF, Nafziger AN, Bertino AJ. Pharmacogenetics affects dosing, efficacy, and toxicity of cytochrome P450 - metabolized drugs. Am J Med 2002; 113 : 746 – 750.
20. Slanař O. Genetický polymorfismus metabolismu léčiv. Postgraduální medicína 2002; 4 : 324 – 330.
21. Kousalová L, Baranová J, Anzenbacher P. Lékové interakce na úrovni cytochromů P450 – část 1. Interakce na úrovni CYP 3A4. Klin Farmakol Farm 2003; 17 : 151 – 157.
22. Špičák J. Léčba inhibitory protonové pumpy. Kardiol Rev Int Med 2014; 16 : 214 – 218.
23. Špinarová L, Vítovec J. Imunosupresivní léčba po transplantaci srdce. Kardiol Rev Int Med 2009; 11 : 63 – 65.
24. Souhrn údajů o přípravku Warfarin Orion 3 a 5 mg. [online] Dostupné z: http:/ / www.sukl.cz/ modules/ medication.
25. Kessler P. Porovnání warfarinu a nových antitrombotik z hlediska lékových interakcí. Klin Farmakol Farm 2012; 26 : 74 – 78.
26. Suchý D, Poklopová Z. Lékové interakce warfarinu s kardiovaskulárními léky. Klin Farmakol Farm 2005; 19 : 41 – 42.
27. Holý J, Havránek P. Zvýšení antikoagulačního účinku warfarinu s krvácivou komplikací vlivem interakce s fluvastatinem – kazuistika. Postgraduální medicína 2008; 10 : 860 – 862.
28. Holý J, Havránek P. Krvácení do trávicího traktu v důsledku lékové interakce warfarinu s ciprofloxacinem – kazuistika. Klin Farmakol Farm 2011; 25 : 92 – 94.
29. Lam MS, Ignoffo RJ, Meibohm B. Klinicky relevantní lékové interakce v onkologii. J Oncol Pharm Pract 2003; 9 : 45 – 85. doi: 10.1191/ 1078155203jp107oa.
30. Zieglmeier M, Hein T. Lékové interakce. Farmakoterapie v klinické praxi. 1st ed. Triton: Praha 2006.
Štítky
Dětská kardiologie Interní lékařství Kardiochirurgie KardiologieČlánek vyšel v časopise
Kardiologická revue – Interní medicína

2015 Číslo 1
Nejčtenější v tomto čísle
- TDM digoxinu v klinické praxi
- Lékové interakce a současná klinická praxe
- Srdeční resynchronizační terapie – kdy a u koho ji v současnosti indikovat?
- TDM antibiotik v klinické praxi