
Plicní arteriální hypertenze
Pulmonary arterial hypertension
Pulmonary arterial hypertension (PAH) is a primary disorder of pulmonary arterioles, either of an unknown aetiology (idiopathic and hereditary PAH) or associated with a known cause (systemic connective tissue disorders, liver disease, congenital cardiac shunts, HIV infection, abuse of some anorectic agents). From the haemodynamic perspective, PAH is characterised as a progressive pre‑capillary pulmonary hypertension leading to relatively rapid right ventricular failure and death. The core examination enabling diagnosis of PAH is Doppler echocardiography. Patients with increased risk of PAH must be regularly monitored with echocardiography. Definitive diagnosis must, however, be made during right heart catheterization. Delayed diagnosis of PAH remains a problem. Non‑specific symptomatology and its late manifestation are the main reasons. The acute vasodilatator test determines pharmacotherapy of PAH. Only the patients with the positive test are indicated for treatment with high doses of calcium channel blockers. This therapy is, together with anticoagulation therapy and heart failure treatment, known as conventional therapy. So‑called specific pharmacotherapy (prostanoids, endothelin receptor antagonists, phosphodiesterase type 5 inhibitors) is, in addition to chronic anticoagulation treatment, indicated in test‑negative patients. Balloon atrial septostomy and lung transplantation are indicated in patients in whom all pharmacotherapeutic options had been attempted.
Keywords:
pulmonary arterial hypertension – prostanoids – endothelin receptor antagonists – phosphodiesterase type 5 inhibitors
Autoři:
P. Jansa; D. Ambrož; P. Poláček; J. Marešová; M. Aschermann; A. Linhart
Působiště autorů:
II. interní klinika kardiologie a angiologie, Centrum pro plicní hypertenzi 1. LF UK a VFN Praha
Vyšlo v časopise:
Kardiol Rev Int Med 2009, 11(4): 166-169
Kategorie:
Sympozium o primárních a nejčastějších sekundárních plicních hypertenzích
Souhrn
Plicní arteriální hypertenze (PAH) je primární onemocnění plicních arteriol, které vzniká buď z neznámé příčiny (idiopatická a hereditární PAH), nebo je jeho vznik asociován se známou vyvolávající příčinou (systémová onemocnění pojiva, jaterní onemocnění, vrozené zkratové srdeční vady, HIV infekce, abúzus některých anorektik). Hemodynamicky je PAH charakterizována jako progredující prekapilární plicní hypertenze vedoucí bez léčby relativně rychle k selhání pravé komory srdeční a ke smrti. Základním vyšetřením sloužícím k detekci PAH je echokardiografie s dopplerovským vyšetřením. Nemocné se zvýšeným rizikem vzniku PAH je nutno pravidelně echokardiograficky vyšetřovat. Definitivní diagnózu je však nutno stanovit při pravostranné srdeční katetrizaci. Velkým problémem zůstává pozdní diagnóza PAH. Příčinou je zejména nespecifická a pozdě se manifestující symptomatologie. O způsobu farmakoterapie PAH rozhoduje test akutní plicní vazodilatace. Pouze nemocní s pozitivním testem jsou indikováni k léčbě vysokými dávkami blokátorů kalciových kanálů. Tato terapie se spolu s antikoagulační léčbou a léčbou srdečního selhání označuje jako léčba konvenční. V případě negativního testu je vedle chronické antikoagulační léčby indikována tzv. specifická farmakoterapie (prostanoidy, antagonisté endotelinových receptorů, inhibitory fosfodiesterázy 5) jako monoterapie nebo jako kombinační léčba. V případě vyčerpání možností farmakoterapie je indikována balónková atriální septostomie a transplantace plic.
Klíčová slova:
plicní arteriální hypertenze – prostanoidy – antagonisté endotelinových receptorů – inhibitory fosfodiesterázy 5
Úvod
Plicní arteriální hypertenze (PAH) je chronické, progresivní a potenciálně fatální onemocnění plicního oběhu vedoucí k selhání pravé komory srdeční, které je způsobeno řadou změn v cévní stěně a v mikrocirkulaci v oblasti plicních arteriol.
V modifikované klinické klasifikaci chronické plicní hypertenze představuje PAH první skupinu vzájemně se patofyziologicky a terapeuticky podobajících klinických stavů. Do dalších skupin patří plicní hypertenze při postižení venul a/nebo plicních kapilár, plicní hypertenze při srdečních onemocněních, plicní hypertenze při respiračních onemocněních, chronická tromboembolická plicní hypertenze a plicní hypertenze z neznámých příčin nebo multifaktoriálního původu (tab. 1) [1]. Do skupiny PAH je řazena především idiopatická a hereditární PAH a dále řada stavů asociovaných se známou vyvolávající příčinou (systémová onemocnění pojiva, jaterní onemocnění, vrozené zkratové srdeční vady, HIV infekce, užívání některých anorektik a další).

Hemodynamicky je PAH definována jako prekapilární plicní hypertenze (střední tlak v plicnici nad 25 mmHg, normální tlak v zaklínění, plicní cévní rezistence nad 3 Woodovy jednotky) [2].
PAH trpí na celém světě nepochybně několik milionů obyvatel. Většina případů však, zejména v méně ekonomicky rozvinutých zemích, uniká diagnóze. V Evropě, USA, Kanadě a Japonsku se výskyt PAH odhaduje celkem na několik set tisíc případů. Podle údajů z francouzského národního registru je minimální prevalence PAH 15 nemocných na milion dospělých obyvatel, zhruba 43% představují nemocní s idiopatickou a hereditární PAH [3]. Nejvyšší výskyt PAH je ve věku 41–60 let, překvapivě častěji u mužů.
Údaje o prognóze specificky neléčené idiopatické PAH pocházejí z údajů amerického NIH registru [4]. Ze souboru 194 nemocných zařazených do registru v letech 1981 až 1985 a sledovaných do roku 1988 přežilo jeden rok 68%, dva roky 48% a tři roky 34% nemocných. Medián přežití byl 2,8 let. Medián přežití u některých asociovaných forem PAH (PAH u systémové sklerodermie, PAH při HIV infekci) je bez specifické léčby ještě výrazně kratší. Naopak lepší prognózu než u idiopatické PAH pozorujeme u nemocných s PAH asociovanou s vrozenou srdeční vadou. Zásadní změnou v osudu nemocných s PAH přineslo až zavedení tzv. specifické léčby s dokumentovanou účinností i po ztrátě vazoreaktivity, tedy v pokročilejších stadiích onemocnění, kdy většinu našich nemocných diagnostikujeme. Do klinické praxe byl nejprve zaveden syntetický analog prostacyklinu – epoprostenol v polovině 90. let 20. století a posléze další analoga prostacyklinu, antagonisté receptorů pro endothelin a inhibitory fosfodiesterázy 5.
Patofyziologie
Genetika
Na rozvoji PAH se podílí kombinace faktorů zevních a genetických. Familiární výskyt PAH je znám desetiletí. Na sklonku minulého století se podařilo prokázat genetickou vazbu onemocnění s markery na chromozomu 2q31–32 [5–6]. Analýza jednoho z kandidátních genů v tomto genomovém regionu prokázala u postižených probandů mutace v genu BMPR2 (bone morphogenetic protein receptor 2), který patří do rodiny receptorů pro TGF beta (transforming growth factor beta). Většina dokumentovaných mutací vede k předčasné terminaci translace proteinu a tím k poruše jeho funkce. U zdravých jedinců BMP inhibuje proliferaci a indukuje apoptózu buněk hladkého svalstva ve stěně plicních arteriol a na buňky endoteliální má účinek antiapoptotický. U nemocných s PAH má BMP na buňky hladkého svalstva účinek proliferativní a proapoptotický účinek je zesílen. Antiapoptotický účinek na buňky endoteliální se mění rovněž v proapoptotický. V případě familiárního výskytu PAH se jedná o onemocnění přenášené autozomálně dominantně s variabilní penetrancí (jen 10–20% nositelů mutace má projevy onemocnění) a expresivitou. Ženy jsou postiženy častěji. Zdá se, že je přítomna také tzv. genetická anticipace, kdy u každé následující postižené generace nastává postižení dříve a/nebo je klinicky závažnější. U idiopatické PAH může být přítomna mutace BMPR2 přibližně u 10–20% pacientů a v tomto případě se pravděpodobně jedná o nové mutace, které mohou být dále předány potomkům. Také ve skupině pacientů s PAH, kteří anamnesticky užívali některá anorektika, byly nalezeny mutace v genu BMPR2 asi u 10% nemocných.
U pacientů s hereditární hemoragickou teleangiektázií (HHT) byla identifikována mutace v genu ALK1 (activin receptor like kinase 1), který tyto pacienty kromě projevů HHT vystavuje riziku vzniku PAH [7].
U nemocných s idiopatickou PAH je také v 65% případů přítomna v homozygotní konstituci varianta polymorfizmu v oblasti promotoru genu pro serotoninový transporter, jehož zvýšená aktivita souvisí s hyperplazií buněk hladkého svalstva ve stěně plicních arteriol.
Celulární mechanizmy vzniku PAH
Hlavní změny v oblasti plicní mikrocirkulace u pacientů s PAH zahrnují vazokonstrikci, cévní remodelaci, trombózu a zánětlivé změny v důsledku relativní nadprodukce faktorů s účinky vazokonstrikčními, růstovými a trombogenními nad faktory s účinky vazodilatačními, antiproliferačními a antitrombotickými.
Prostacyklin a tromboxan A2 jsou hlavní metabolity kyseliny arachidonové. Prostacyklin je silný vazodilatátor, inhibitor aktivace trombocytů a má rovněž významné účinky antiproliferační. Tomboxan A2 je naproti tomu potentní vazokonstriktor a destičkový agonista. U nemocných s PAH je metabolizmus posunut ve prospěch tromboxanu.
U PAH je dokumentována zvýšená plazmatická koncentrace vazokonstrikčního endotelinu 1 a serotoninu, naopak bývá snížená aktivita endoteliální NO syntázy a koncentrace dalšího vazodilatačního faktoru, vazoaktivního intestinálního peptidu.
V patofyziologii PAH hrají důležitou roli draslíkové kanály. Inhibice voltážově řízených draslíkových kanálů (Kv), např. hypoxií a deriváty fenfluraminu, nebo jejich downregulace vede k depolarizaci buněčné membrány a k influxu vápenatých iontů do cytosolu. V trombocytech dochází ke zvýšení efluxu serotoninu a k inhibici jeho zpětného vychytávání. Důsledkem je vazokonstrikce a proliferace.
Pro podíl zánětu na patofyziologii PAH svědčí v řadě případů nalézaná pozitivita autoprotilátek, infiltrace cévních lézí zánětlivými elementy a depozita komplementu ve stěně plicních cév.
U PAH je známa řada abnormit koagulačního a fibrinolytického systému vedoucí ke zvýšené pohotovosti ke vzniku trombů.
Diagnostika
Při manifestaci symptomů budících podezření na PAH (dušnost, únavnost, bolesti na hrudi, synkopy) nebo při snaze detekovat PAH v rámci screeningových programů (tab. 2) je klíčovým neinvazivním vyšetřením echokardiografie [8–9]. Systolický tlak v plicnici lze odhadnout z vrcholového gradientu trikuspidální regurgitace po přičtení tlaku v pravé síni. Plicní hypertenze je nepravděpodobná, pokud je odhadovaný systolický tlak v plicnici menší než 36 mmHg a nejsou přítomny další známky, které mohou svědčit pro plicní hypertenzi (například dilatace a hypertrofie pravé komory, D tvar levé komory, případně zkrácení akceleračního času proudění v plicnici). Plicní hypertenze je možná, pokud odhadovaný systolický tlak v plicnici dosahuje hodnot 37–50 mmHg. Za pravděpodobnou považujeme plicní hypertenzi při echokardiografickém odhadu tlaku v plicnici nad 50 mmHg.


K odlišení jiných příčin plicní hypertenze slouží řada dalších vyšetřovacích metod. Funkční vyšetření plic je nezbytné k vyloučení významného respiračního onemocnění jako příčiny plicní hypertenze, polysomnografie k vyloučení syndromu obstrukční spánkové apnoe. Ventilační a perfuzní scintigrafie plic, případně CT angiografie je nezbytná k odlišení chronické tromboembolické plicní hypertenze.
Definitivní diagnóza PAH je katetrizační. Pravostranná srdeční katetrizace kromě hemodynamického vyšetření umožňuje provedení testu akutní plicní vazodilatace. Ten je indikován zejména u nemocných s idiopatickou a hereditární PAH a u nemocných s PAH v důsledku abúzu anorektik. Za kritéria pozitivity testu se považuje pokles středního tlaku v plicnici alespoň o 10 mmHg vstupních hodnot a zároveň pod 40 mmHg bez současného zhoršení srdečního výdeje. Testování vazoreaktivity má být prováděno zásadně v expertním centru pro PAH.
K určení pokročilosti onemocnění se využívá jednoduchý funkční test – šestiminutová chůze a také vyšetření některých biomarkerů: kyselina močová, troponin, natriuretické peptidy. Výsledky těchto vyšetření umožňují spolu s dalšími klinickými, echokardiografickými a hemodynamickými daty stratifikovat nemocné a tak individualizovat léčbu.
Léčba
Režimová opatření
Fyzická zátěž je u nemocných s PAH vhodná dle individuální tolerance, optimální je lehké aerobní cvičení (chůze). Při cestě letadlem je nezbytná inhalace kyslíku. Doporučováno je očkování proti chřipce. Těhotenství je u PAH kontraindikováno, nutná je účinná antikoncepce. Z hlediska rizika tromboembolizmu je akceptovatelná hormonální antikoncepce při současné antikoagulační léčbě.
Konvenční léčba
Ke konvenčním léčebným možnostem u PAH patří diuretika, oxygenoterapie, antikoagulace – a léčba vazodilatačními blokátory kalciových kanálů. Léčba blokátory kalciových kanálů je indikována pouze v případě pozitivního vazodilatačního testu. Dlouhodobá dobrá klinická odpověď je charakteristická mj. zlepšením symptomů do stadia NYHA I–II a vyžaduje opakovaného ověření v intervalu několika měsíců. Po 3–4 měsících od zahájení léčby má být její efekt také ověřen hemodynamickým vyšetřením [10]. Při selhání léčby blokátory kalciových kanálů je nezbytná specifická farmakoterapie.
Specifická farmakoterapie PAH
Většina nemocných s PAH nesplňuje kritéria vazoreaktivity a jsou indikováni ke specifické farmakoterapii [11] (obr. 1). Její indikace vyžaduje přísně individuální přístup. U symptomatických nemocných má být léčba zahájena bezprostředně po stanovení diagnózy. Cílem je u pacientů ve funkční třídě NYHA II zabránit progresi onemocnění, u nemocných ve funkční třídě NYHA III a IV dosáhnout zlepšení.
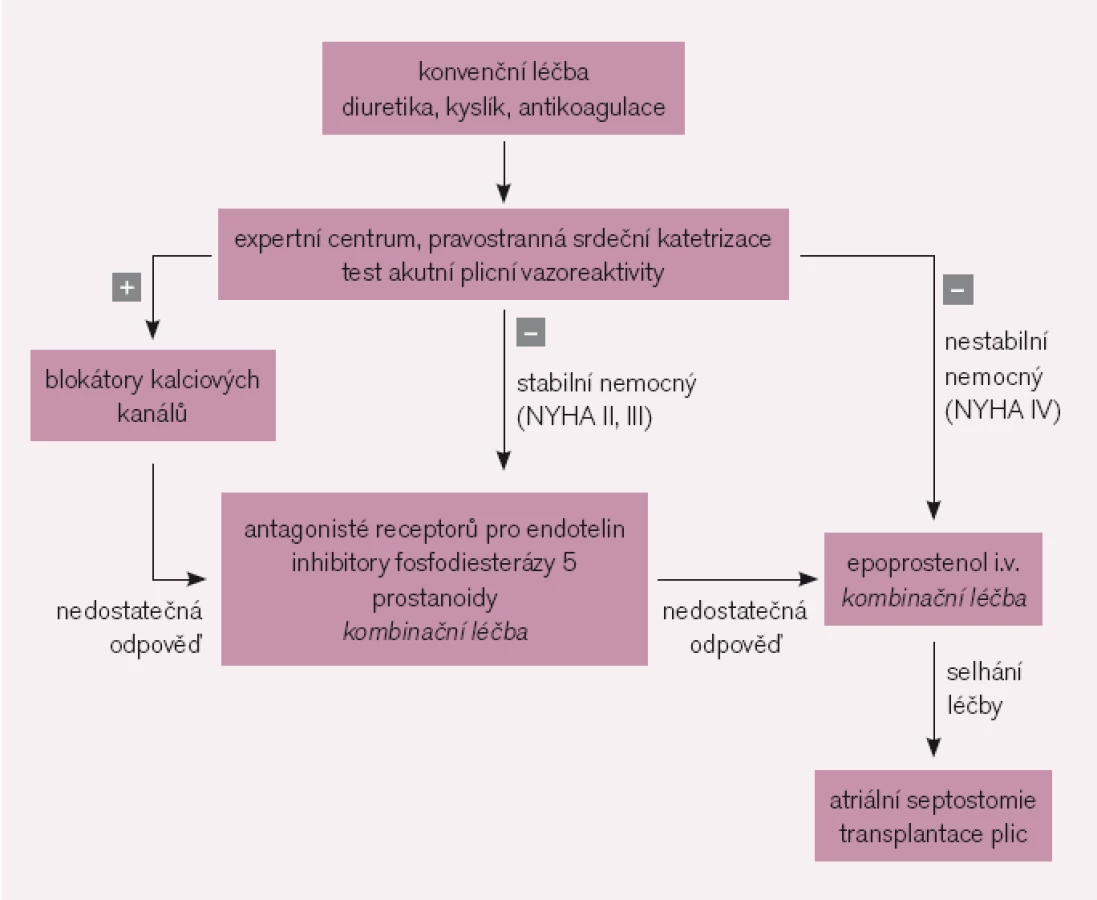
U pacientů ve funkčním stadiu NYHA II je metodou volby perorální léčba, kterou ve většině případů zahajujeme antagonisty receptorů pro endotelin nebo inhibitory fosfodiesterázy 5. U nemocných ve funkčním stadiu NYHA III je indikována léčba antagonisty receptorů pro endotelin nebo inhibitory fosfodiesterázy 5, případně prostanoidy. Ve funkčním stadiu NYHA IV je základním lékem intravenózní prostacyklin. V případě nedostatečné odpovědi na léčbu jedním přípravkem je indikována sekvenční kombinační léčba.
Nefarmakologická léčba PAH
Balónková atriální septostomie spočívá ve vytvoření umělé komunikace na úrovni síní se vznikem pravolevého zkratu [12]. Cílem intervence je zvýšení srdečního výdeje za cenu systémové desaturace. Může sloužit jako most k transplantaci u nemocných s refrakterním pravostranným srdečním selháním a synkopami. V zemích, kde specifická léčba PAH není k dispozici, je atriální septostomie často jedinou možnou terapeutickou intervencí.
Transplantace plic představuje účinnou léčbu u nemocných v terminálním stadiu PAH po vyčerpání všech ostatních dostupných léčebných možností. Nemocní ve funkčním stadiu NYHA IV mají být zařazeni na čekací listinu k transplantaci plic ihned po stanovení diagnózy a mohou být vyřazeni při zlepšení do funkčního stadia NYHA II. Nemocní ve funkčním stadiu NYHA III jsou indikováni k transplantaci, pokud ani kombinační léčba PAH nevede k výraznějšímu zlepšení. Jednoroční přežití po transplantaci plic pro PAH se pohybuje mezi 66% a 75%.
Závěr
PAH zůstává nadále nevyléčitelným onemocněním, na jehož patofyziologii se podílí porucha v řadě regulačních mechanizmů. Díky specifické léčbě PAH nemocní s tímto onemocněním žijí v posledních dvou desetiletích jistě kvalitněji a také déle. Velké naděje se vkládají nejen do kombinačních schémat a nových forem podávání již známých léků, ale také do vývoje nových přípravků. V současné době je v různém stadiu klinického testování zhruba 10 perspektivních léků. Část z nich jistě změní algoritmus léčby v průběhu příštího desetiletí. Stále neuspokojivou prognózu řady nemocných však lze ovlivnit již nyní správně a včas stanovenou diagnózou a také časnou terapeutickou intervencí na pracovišti s dostatečnou zkušeností a bohatým multidisciplinárním zázemím.
Doručeno do redakce 4. 10. 2009
Přijato po recenzi 25. 11. 2009
MUDr. Pavel Jansa
MUDr. David Ambrož
MUDr. Pavel Poláček
MUDr. Jana Marešová
prof. MUDr. Michael Aschermann, DrSc.
prof. MUDr. Aleš Linhart, DrSc.
II. interní klinika kardiologie a angiologie,
Centrum pro plicní hypertenzi 1. LF UK a VFN Praha
jansapavel@yahoo.com
Zdroje
1. Simonneau G, Robbins IM, Beghetti M et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2009; 54: S43–S54.
2. Galie N, Hoeper MM, Humbert M et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. The task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Respir J 2009 Sep 12 [Epub ahead of print].
3. Humbert M, Sitbon O, Chaouat A et al. Pulmonary arterial hypertension in France. Results from a national registry. Am J Respir Crit Care Med 2006; 173 : 1023–1030.
4. D’Alonzo GE, Barst RJ, Ayres SM et al. Survival in patients with primary pulmonary hypertension: results from a national prospective registry. Ann Intern Med 1991; 115 : 343–349.
5. Nichols WC, Koller DL, Slovis B et al. Localization of the gene for familial primary pulmonary hypertension to chromosome 2q31–32. Nat Genet 1997; 15 : 277–80.
6. Morse JH, Jones AC, Barst RJ et al. Mapping of familial primary pulmonary hypertension locus (PPH1) to chromosome 2q31–q32. Circulation 1997; 95 : 2603–2606.
7. Trembath RC, Thomson JR, Machado RD et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2001; 345 : 325–334.
8. Daniels LB, Krummen DE, Blanchard DG. Echocardiography in pulmonary vascular disease. Cardiol Clin 2004; 22 : 383–399.
9. McGoon MD, Garvan CK. Pulmonary hypertension: diagnosis and management. Mayo Clin Proc 2009; 84 (2): 191–207.
10. Sitbon O, Humbert M, Jais X et al. Long‑term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 2005; 111 : 3105–3111.
11. Barst RJ, Gibbs JS, Ghofrani HA et al. Updated evidence‑based treatment algorithm in pulmonary arterial hypertension. J Am Coll Cardiol 2009; 54: S78–S84.
12. Keogh AM, Mayer E, Benza RL et al. Interventional and surgical modalities of treatment in pulmonary hypertension. J Am Coll Cardiol 2009; 54: S67–S77.
Štítky
Dětská kardiologie Interní lékařství Kardiochirurgie KardiologieČlánek vyšel v časopise
Kardiologická revue – Interní medicína

2009 Číslo 4
Nejčtenější v tomto čísle
- Pleurální výpotky – etiologie a diagnostika
- Plicní embolizace
- Vrozené srdeční vady s plicní hypertenzí
- Plicní hypertenze u chronické obstrukční plicní nemoci