
Ischemicko-reperfuzní poškození po srdeční zástavě a protektivní účinky hypotermie
Ischemia-reperfusion injury following cardiac arrest and protective effects of hypothermia
Ischemia-reperfusion injury following cardiac arrest and protective effects of hypothermia. Early restoration of spontaneous circulation after successful cardiopulmonary resuscitation is an essential lifesaving step. However, within minutes of global ischemia, several pathways are triggered that progress even after reperfusion and may cause further damage to ischemic but viable cells (ischemia-reperfusion (IR) injury). It is desirable, therefore, besides general resuscitation care and treatment of the cause of cardiac arrest to prevent also the harmful effects of IR injury. Several pathogenic mechanisms participate in IR injury: reactive oxygen species, ion imbalance, proteases activation, induction of apoptosis, and activation of inflammation. The majority of these processes are not tissue specific and may be detectable in different organs. In ischemia very sensitive brain tissue, IR injury is enhanced by accumulation and excitotoxicity of some neurotransmiters, disruptions of bloodbrain barrier, or oedema. Together with increasing knowledge of IR mechanism, substantial efforts have been made to develop protective methods against IR injury. Many therapeutic interventions targeted against “key” mechanisms of IR injury showed very promising results in experimental studies but failed completely in clinical settings. Recently, however, one therapeutic approach has been proved to be very effective also in clinical trials: mild hypothermia. Experimental studies have shown that hypothermia exerts beneficial effects simultaneously on various parallel pathways participating in IR injury and leads to significant neuroprotection. Therapeutic hypothermia, therefore, became one of the crucial methods in the current management of patients after cardiac arrest.
Keywords:
cardiac arrest – cardiopulmonary resuscitation – ischemia-reperfusion injury – hypothermia
Autoři:
P. Ošťádal
Působiště autorů:
Kardiocentrum, kardiologické oddělení Nemocnice Na Homolce, Praha
Vyšlo v časopise:
Kardiol Rev Int Med 2009, 11(1): 11-15
Souhrn
Časné obnovení krevního oběhu při úspěšné kardiopulmonální resuscitaci pro srdeční zástavu znamená zásadní krok k záchraně života. Během minuty až desítek minut trvající globální ischemii se však spouští řada dějů, které probíhají i po obnovení cirkulace (v reperfuzi) a jež mohou vést k dalšímu poškození dosud viabilních buněk (ischemicko‑reperfuzní (IR) poškození). Je proto žádoucí se kromě všeobecné resuscitační péče a léčby příčiny srdeční zástavy pokusit také o omezení negativních dějů provázejících reperfuzi. Na poškození buněk v reperfuzi se zřejmě podílejí volné kyslíkové radikály, iontová dysbalance, aktivace proteáz, indukce apoptózy a významnou roli hraje jistě také aktivace zánětlivých mechanizmů. Většina těchto procesů není tkáňově specifická a může probíhat v různých orgánech. V mozku, k ischemii velmi citlivé tkáni, se na IR poškození podílí i akumulace škodlivých neurotransmiterů, změny propustnosti hematoencefalické bariéry či edém. Spolu s prohlubující se znalostí patogenetických mechanizmů odpovědných za vznik IR poškození se opakovaně objevovaly snahy o protektivní ovlivnění reperfundované tkáně. Byla popsána řada intervencí zaměřených na „klíčové“ faktory IR poškození, které v experimentu často vedly až k zázračným výsledkům, v klinických studiích však většinou zcela selhaly. Jeden terapeutický postup se však z této neúspěšné řady vymyká – řízená hypotermie; v experimentech bylo prokázáno, že hypotermie pravděpodobně pozitivně ovlivňuje většinu z výše popsaných patogenetických faktorů; detailní mechanizmy tohoto účinku však dosud známy nejsou. První klinické studie ukázaly, že tento postup přináší jednoznačný prospěch, především v protekci proti poškození mozku; řízená hypotermie je proto dnes jedním ze zásadních pilířů léčby nemocných po resuscitaci pro srdeční zástavu.
Klíčová slova:
srdeční zástava – kardiopulmonální resuscitace – ischemicko‑reperfuzní poškození – hypotermie
Úvod
Úspěšná kardiopulmonální resuscitace a obnovení spontánní cirkulace po srdeční zástavě je zcela nezbytným krokem k záchraně života; povšechná ischemie během oběhové zástavy však vede k aktivaci celé řady patofyziologických procesů, postihujících celý organizmus, které mohou způsobit další, často fatální poškození. Tento stav bývá označován jako „postresuscitační nemoc“ [1] nebo nověji jako „post cardiac arrest syndrome (PCAS)“ [2–3].
Ischemicko-reperfuzní poškození po srdeční zástavě
Ačkoli patofyziologické pochody, podílející se na PCAS, spouštěné ischemií a reperfuzí, jsou do značné míry podobné v různých tkáních, z hlediska klinické manifestace je můžeme rozdělit na: (i) poškození mozku, (ii) dysfunkce myokardu, (iii) systémová ischemicko reperfuzní reakce a (iv) perzistující základní onemocnění [2–3]. Klinická závažnost těchto projevů závisí na celé řadě faktorů, především na stavu organizmu před oběhovou zástavou, přítomných komorbiditách a délce trvání povšechné ischemie. Pro další prognózu pacienta je zcela zásadní ovlivnění základního onemocnění, se snahou zabránit opakování oběhové zástavy, hemodynamická stabilizace, zajištění dostatečné oxygenace, normalizace vnitřního prostředí a jak se v poslední době stále více ukazuje – neuroprotekce. Podle některých údajů je totiž poškození mozku zodpovědné až za 68% úmrtí nemocných přijatých na jednotku intenzivní péče po resuscitaci pro srdeční zástavu mimo nemocnici [4].
Příznivé ovlivnění ischemicko reperfuzního poškození po srdeční zástavě je jedním z klíčových postupů, které mohou významným způsobem ovlivnit jak samotné přežití, tak i kvalitu života nemocného. Je proto pochopitelné, že mechanizmy PCAS a možnosti jejich ovlivnění jsou v současné době předmětem intenzivního zájmu řady experimentálních i klinických laboratoří.
Poškození mozku
Mechanizmus mozkového poškození po srdeční zástavě zahrnuje celou řadu patogenetických pochodů, od excitotoxického působení některých neuromediátorů přes porušenou homeostázu iontů, především vápníku, uvolnění volných radikálů, patologickou aktivaci proteáz až k aktivaci apoptózy (obr. 1).
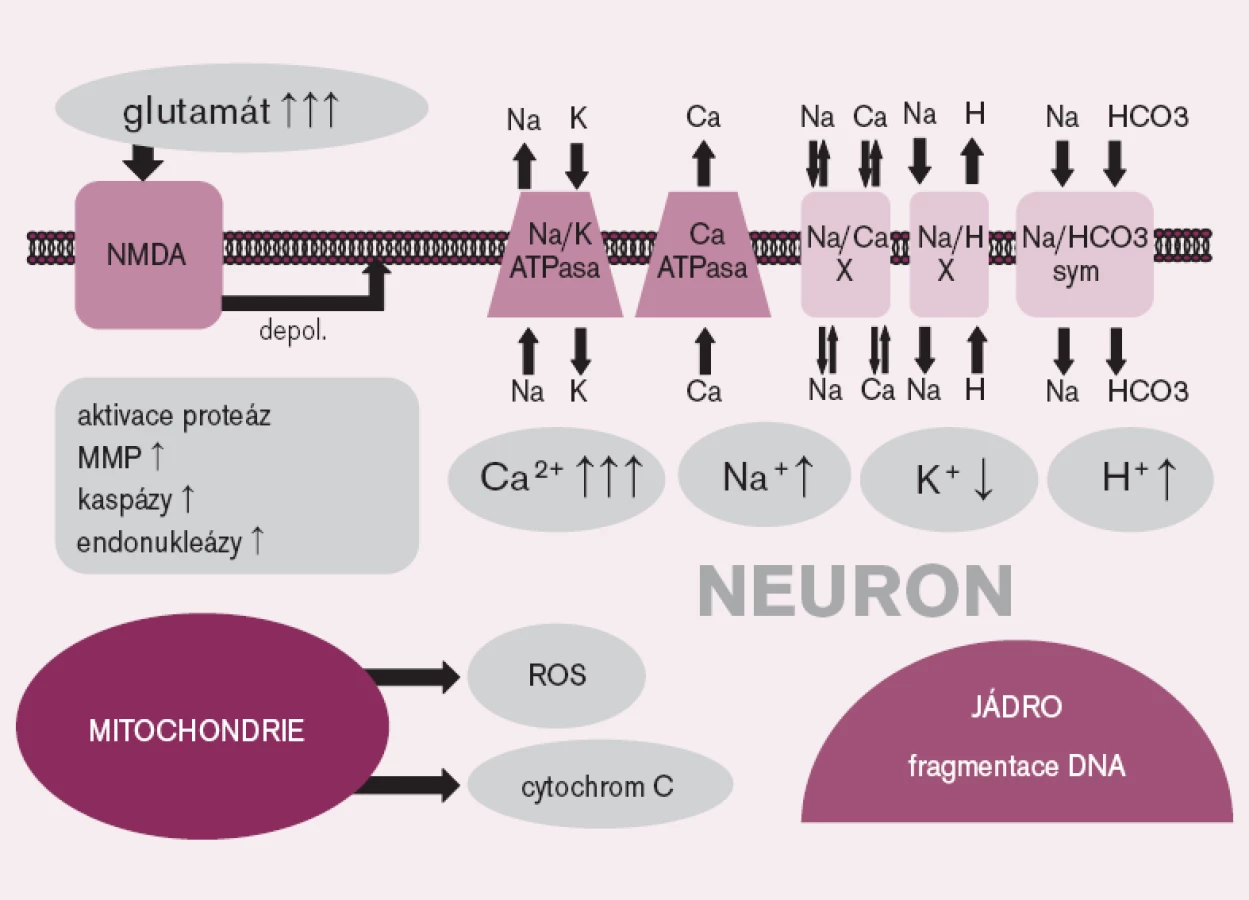
V průběhu PCAS byl popsán zánik mozkových buněk jak nekrózou, tak apoptózou; relativní podíl obou procesů se podle různých autorů liší a závisí zřejmě na řadě dalších faktorů [5]. Na poškození mozku se účastní mikrovaskulární postižení tzv. „no-reflow“ fenoménem; mechanizmem, který není doposud zcela vysvětlen [6]. Podílí se na něm pravděpodobně intravaskulární trombóza, která vede k poruchám perfuze a k pokračování regionální ischemie i po obnovení krevního průtoku mozkem [7]. U nemocných po srdeční zástavě bývá také popisován edém mozku, jen zřídka provázený zvýšením nitrolebního tlaku; není však jasné, zda edém může přispívat ke zhoršení perfuze nebo zda se jedná pouze o následek předchozího ischemického poškození [8]. Dalším nepříznivým patogenetickým faktorem je relativně často pozorovaná hypertermie. Vzestup tělesné teploty nad 37°C je spojen s horší prognózou včetně zvýšené mortality [9–10].
V průběhu mozkové ischemie dochází také k synaptickému hromadění glutamátu – aminokyseliny, která působí jako neurotransmiter, a to především kvůli selhání energeticky náročného zpětného vstřebávání tohoto mediátoru do glií. Glutamát ve vysoké koncentraci aktivuje glutamátové receptory (především N-methyl-D-aspartát (NMDA) receptory), což způsobuje protrahovanou depolarizaci nervových buněk, vedoucí k influxu iontů vápníku intracelulárně s následnou iontovou nerovnováhou a stimulací dalších, buňku poškozujících procesů, jako je aktivace proteáz [11].
V průběhu ischemie a reperfuze se z poškozených mitochondrií uvolňují ve zvýšeném množství reaktivní formy kyslíku, které spolu s ischemickým poškozením antioxidačních obranných systémů způsobují oxidativní stres [12]. Nadbytek reaktivních forem kyslíku vede k poškození buňky na nejrůznějších úrovních – dochází např. k peroxidaci lipidů, přímému poškození DNA či k aktivaci apoptózy [11].
V ischemickém mozku byl opakovaně prokázán zánik buněk mechanizmem apoptózy, která je spouštěna buď vnitřní, na mitochondriích závislou cestou, nebo aktivací specifických receptorů. Vnitřní cesta aktivace apoptózy je zahájena uvolněním cytochromu-c z mitochondrií s následnou aktivací kaspázy 9, která aktivuje kaspázu 3, a ta štěpí jadernou DNA. Vnější, receptorová cesta, začíná navázáním pro apoptotického ligandu na specifický receptor (například Fas/FasL), čímž se aktivuje kaspáza 8 a následně kaspáza 3 [11].
V průběhu protrahované mozkové ischemie dochází také ke spuštění zánětlivých mechanizmů – aktivují se mikroglie, zvyšuje se exprese endoteliálních adhezivních molekul a dochází k infiltraci poškozené tkáně periferními leukocyty [13]. Imunitní buňky potom uvolňují celou řadu faktorů vedoucích k amplifikaci zánětu a k dalšímu poškození tkáně (např. oxid dusnatý, prozánětlivé cytokiny, superoxid a proteázy) [11].
Ischemie také poškozuje hematoencefalickou bariéru, vede k disrupci endoteliálních „tight juntions“ a ke zvýšení cévní permeability [14–15]. Tyto změny přispívají k prostupu tekutiny do intersticiálního prostoru, a tím ke vzniku edému mozku.
Dysfunkce myokardu
S poruchou systolické funkce myokardu po srdeční zástavě se setkáváme často a lze předpokládat, že snížený minutový srdeční výdej jako její důsledek přispívá ke špatné prognóze nemocných po úspěšné resuscitaci [4]. Příčinou globální dysfunkce myokardu, přetrvávající po obnovení krevního oběhu, je pravděpodobně omráčení (stunning) myokardu, a není proto překvapivé, že porucha kontraktility je zpravidla pouze přechodná a přetrvává pouze hodiny až dny, výjimečně i měsíce [16–17]. Bylo opakovaně popsáno, že omráčený myokard po srdeční zástavě reaguje velmi dobře na inotropní podporu [12,18].
Systémová ischemicko reperfuzní reakce
Srdeční zástava představuje nejtěžší formu šokového stavu, během které je zastavena dodávka kyslíku a metabolických substrátů tkáním. Úspěšná resuscitace zlepšuje tento stav jen částečně: dochází sice k obnově krevního oběhu, ale minutový srdeční výdej je výrazně nižší než za normálních okolností. V důsledku dysfunkce myokardu, hemodynamické nestability a případného selhání mikrocirkulace tak pokračuje neadekvátní zásobení tkání kyslíkem [2]. Protrahovaný kyslíkový dluh vede k aktivaci endotelu a spuštění systémové zánětlivé reakce, zvyšuje se riziko multiorgánového selhání a infekce [19]. Vzestup sérových koncentrací solubilní intercelulární adhezivní molekuly 1 (sICAM 1), vaskulární buněčné adhezivní molekuly 1 (VCAM 1) a selektinu E a P, které bývají nacházeny po úspěšné resuscitaci, nepřímo ukazuje na aktivaci leukocytů a poškození endotelu [20–21].
Po resuscitaci navíc dochází k narušení rovnováhy koagulačního systému. Aktivace prokoagulačních mechanizmů, která není provázena dostatečnou aktivací fibrinolýzy, se může podílet na selhání mikrocirkulace intravaskulární tvorbou fibrinu a vznikem mikrotrombů [22]. Bývá též pozorována relativní adrenální dysfunkce – nedostatečná odpověď nadledvin na stimulaci kortikotropním hormonem, přestože hladina kortizolu je po resuscitaci často zvýšená [23].
Perzistující základní onemocnění
Patofyziologii PCAS také komplikuje základní onemocnění, které je přímo odpovědné za srdeční zástavu nebo se na jejím vzniku podílí. Stanovení diagnózy, průběh a léčba akutního onemocnění, jako jsou akutní koronární syndrom (AKS), plicní onemocnění, sepse, krvácení či různé intoxikace, mohou dále komplikovat PCAS a být jeho rozvojem komplikovány.
AKS je příčinou srdeční zástavy mimo nemocnici u přibližně 40–50% dospělých nemocných. Akutní uzávěr koronární tepny byl nalezen u 48% nemocných po srdeční zástavě bez zřejmé nekardiální příčiny [24]. Už během resuscitace bývá u 40% nemocných zvýšená hladina troponinu, ukazující na předcházející akutní infarkt myokardu [25]. Poškození myokardu při protrahované globální ischemii však snižuje specifitu srdečních biomarkerů pro stanovení diagnózy AKS u nemocných po resuscitaci. Za 12 hodin po obnovení krevního oběhu klesá specifita troponinu pro diagnózu infarktu myokardu na 80% [26]. U dospělých nemocných se srdeční zástavou v nemocnici byl AKS diagnostikován jako příčina pouze u 11% [27].
Plicní embolie je odpovědná za 2–10% náhlých úmrtí [27]. Také primární plicní onemocnění jako chronická obstruktivní plicní nemoc, astma nebo pneumonie mohou vést k respiračnímu selhání s následnou srdeční zástavou. Zdá se, že poškození mozku způsobené srdeční zástavou při ventilačním selhání je těžší než při primárně cirkulačním selhání. Je možné, že zásadní škodlivý vliv zde má perfuze hypoxemickou krví během asfyxie [2]. Možnou příčinou srdeční zástavy je také sepse, poškození plic (Acute Respiratory Distress Syndrome – ARDS) a multiorgánové selhání. Sepse se proto může zásadně podílet na zhoršení průběhu PCAS [4].
Protektivní účinky hypotermie
V posledních letech bylo vynaloženo značné úsilí na výzkum ischemicko reperfuzního poškození a možností jeho ovlivnění. Je však třeba zmínit, že většina postupů, které se v experimentu zdály být slibné až zázračné, v klinice zcela selhala. Sem patří např. antioxidanty, inhibitory Na-Ca výměníku nebo protilátky proti neutrofilům a složkám komplementu [12]. Jedním z možných vysvětlení neúspěchu těchto intervencí je, že ovlivňují vždy pouze úzké spektrum patogenetických mechanizmů. Jak bylo popsáno výše, na patogenezi ischemicko reperfuzního poškození se však podílí celá řada navzájem souvisejících pochodů [12].
Ze zmíněných neúspěšných pokusů o ovlivnění ischemicko reperfuzního poškození se vymyká jeden postup, který se ukázal být úspěšný nejenom v experimentálních, ale i v klinických studiích – tímto postupem je navození mírné hypotermie [28]. Dvě randomizované klinické studie a jejich meta analýza ukázaly, že řízená mírná hypotermie vedla ke zlepšení výsledků u dospělých, kteří zůstávali v bezvědomí po resuscitaci pro srdeční zástavu mimo nemocnici [29–31]. Zdá se, že výjimečností hypotermie v ovlivnění ischemicko reperfuzního poškození je její příznivé působení na celou řadu souběžně probíhajících procesů (tab. 1).

Hypotermie a metabolizmus
Hypotermie snižuje buněčný metabolizmus, zmenšuje spotřebu energie a zvyšuje postischemickou utilizaci glukózy [32]. Pokles teploty o 1 °C snižuje úroveň metabolizmu v mozku o 5–7% [33]. Hypotermie redukuje přestup iontů vápníku z extracelulárního prostoru intracelulárně a zlepšuje také homeostázu draslíku [11].
Hypotermie a průtok krve mozkem
Za normálních okolností je průtok krve mozkem přibližně 50 mL/100g/min; s poklesem tělesné teploty se tato hodnota snižuje. Podle experimentální studie Mori a spol. [34] na zvířecím modelu klesá průtok krve mozkem z 48 mL/100g/min při normotermii na 21 mL/100g/min při teplotě 33°C a 11 mL/100g/min při ochlazení na 29°C. Během ischemie a reperfuze je však vztah mezi tělesnou teplotou a průtokem krve mozkem mnohem méně jasný: po obnovení krevního oběhu dochází zpravidla k přechodnému vzestupu průtoku, hyperemii; následně se průtok mozkem postupně snižuje. Zdá se, že hypotermie zabraňuje přechodné hyperemii, a pomáhá naopak udržet průtok v následném období [11].
Hypotermie a neurotransmitery
Neurotransmitery se během ischemie začínají ve zvýšené míře vyplavovat za 10–20 min, jejich hladina dosahuje vrcholu přibližně za 60 min a k normálu se navrací po cca 90–120 min [35]. Mírná hypotermie v průběhu ischemie snižuje uvolňování mediátorů, při teplotě 30–33 °C je zcela inhibováno vyplavení glutamátu [36]. Zmenšení vzestupu hladiny glutamátu je spojeno s poklesem intracelulární koncentrace vápníku, se zmenšením spotřeby adenosin trifosfátu (Adenosine Triphosphate – ATP) a představuje pravděpodobně jeden z klíčových neuroprotektivních účinků hypotermie [11,37].
Hypotermie, oxidativní stres a apoptóza
Hypotermie inhibuje tvorbu volných radikálů, snižuje peroxidaci lipidů a ochraňuje jadernou DNA před přímým poškozením reaktivními formami kyslíku a fragmentací [38–39]. Bylo navíc ukázáno, že hypotermie stimuluje produkci anti apoptotického proteinu Bcl-2, inhibuje uvolnění cytochromu C a aktivaci kaspáz [11,40].
Hypotermie a zánět
Hypotermie působí protizánětlivě, zmenšuje počet neutrofilů v ischemií postižené tkáni a inhibuje aktivaci mikroglií. Hypotermie také snižuje expresi ICAM 1 a ovlivňuje tím interakce mezi leukocyty a endotelem, snižuje produkci různých prozánětlivých mediátorů, jako jsou oxid dusnatý nebo interleukin 6, na čemž se možná podílí inhibice transkripce nukleárního faktoru kappa B (NFkB) [41–43]. Protizánětlivé účinky hypotermie jsou vyjádřeny nejen v přítomnosti nekrózy, ale i tam, kde jsou buňky sice ischemií poškozeny, ale přežívají [43].
Hypotermie a hematoencefalická bariéra, cévní permeabilita a vznik edému
Hypotermie omezuje vznik disrupcí hematoencefalické bariéry a snižuje cévní permeabilitu po ischemicko reperfuzním poškození, čímž zmenšuje riziko vzniku edému [14–15]. Bylo popsáno, že hypotermie snižuje tvorbu matrixových metaloproteináz, které se pravděpodobně významně podílejí na poškození hematoencefalické bariéry během ischemie a reperfuze [44].
Závěr
Ischemicko reperfuzní poškození po srdeční zástavě je vážný klinický problém současné medicíny odpovědný za většinu úmrtí nemocných po úspěšné resuscitaci mimo nemocnici. Po řadě neúspěšných pokusů o ovlivnění tohoto ischemicko reperfuzního poškození byl navržen nový terapeutický postup, který prokazatelně významně zlepšuje prognózu nemocných po srdeční zástavě – řízená mírná hypotermie. Hypotermie příznivě ovlivňuje současně různé paralelně probíhající procesy, které se na patogenezi ischemicko reperfuzního poškození podílejí.
Na základě současných znalostí je však zcela zřejmé, že obnovením spontánního krevního oběhu po srdeční zástavě záchrana života nemocného nekončí, ale začíná.
Práce byla podpořena z Výzkumného záměru 00000064203 Ministerstva zdravotnictví ČR.
Doručeno do redakce 14. 2. 2009
Přijato po recenzi 5. 3. 2009
doc. MUDr. Petr Ošťádal, Ph.D., FESC
Kardiocentrum, kardiologické oddělení
Nemocnice Na Homolce, Praha
petr.ostadal@homolka.cz
Zdroje
1. Negovsky VA, Gurvitch AM. Post‑resuscitation disease – a new nosological entity. Its reality and significance. Resuscitation 1995; 30 : 23–27.
2. Neumar RW, Nolan JP, Adrie C et al. Post‑cardiac arrest syndrome: epidemiology, pathophysiology, treatment, and prognostication. A consensus statement from the International Liaison Committee on Resuscitation (American Heart Association, Australian and New Zealand Council on Resuscitation, European Resuscitation Council, Heart and Stroke Foundation of Canada, InterAmerican Heart Foundation, Resuscitation Council of Asia, and the Resuscitation Council of Southern Africa); the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; and the Stroke Council. Circulation 2008; 118 : 2452–2483.
3. Nolan JP, Neumar RW, Adrie C et al. Post‑cardiac arrest syndrome: epidemiology, pathophysiology, treatment, and prognostication. A Scientific Statement from the International Liaison Committee on Resuscitation; the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; the Council on Stroke. Resuscitation 2008; 79 : 350–379.
4. Laver S, Farrow C, Turner D et al. Mode of death after admission to an intensive care unit following cardiac arrest. Intensive Care Med 2004; 30 : 2126–2128.
5. Martin LJ, Al-Abdulla NA, Brambrink AM et al. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: A perspective on the contributions of apoptosis and necrosis. Brain Res Bull 1998; 46 : 281–309.
6. Wolfson SK Jr, Safar P, Reich H et al. Dynamic heterogeneity of cerebral hypoperfusion after prolonged cardiac arrest in dogs measured by the stable xenon/CT technique: a preliminary study. Resuscitation 1992; 23 : 1–20.
7. Fischer M, Böttiger BW, Popov-Cenic S et al. Thrombolysis using plasminogen activator and heparin reduces cerebral no-reflow after resuscitation from cardiac arrest: an experimental study in the cat. Intensive Care Med 1996; 22 : 1214–1223.
8. Morimoto Y, Kemmotsu O, Kitami K et al. Acute brain swelling after out-of-hospital cardiac arrest: pathogenesis and outcome. Crit Care Med 1993; 21 : 104–110.
9. Zeiner A, Holzer M, Sterz F et al. Hyperthermia after cardiac arrest is associated with an unfavorable neurologic outcome. Arch Intern Med 2001; 161 : 2007–2012.
10. Langhelle A, Tyvold SS, Lexow K et al. In-hospital factors associated with improved outcome after out-of-hospital cardiac arrest. A comparison between four regions in Norway. Resuscitation 2003; 56 : 247–263.
11. Liu L, Yenari MA. Therapeutic hypothermia: neuroprotective mechanisms. Front Biosci 2007; 12 : 816–825.
12. Ostadal P, Zdobnicka I, Dhalla NS. Does Reperfusion Cause Any Injury to the Myocardium? In: Dhalla NS, Chockalingam A, Berkowitz HI et al (eds). Frontiers in Cardiovascular Health. Boston, Dordrecht, London: Kluwer Academic Publishers 2003 : 145–159.
13. Danton GH, Dietrich WD. Inflammatory mechanisms after ischemia and stroke. J Neuropathol Exp Neurol 2003; 62 : 127–136.
14. Chi OZ, Liu X, Weiss HR. Effects of mild hypothermia on blood-brain barrier disruption during isoflurane or pentobarbital anesthesia. Anesthesiology 2001; 95 : 933–938.
15. Jurkovich GJ, Pitt RM, Curreri PW et al. Hypothermia prevents increased capillary permeability following ischemia-reperfusion injury. J Surg Res 1988; 44 : 514–521.
16. Laurent I, Monchi M, Chiche JD et al. Reversible myocardial dysfunction in survivors of out-of-hospital cardiac arrest. J Am Coll Cardiol 2002; 40 : 2110–2116.
17. Ostadal P. What is ‚reperfusion injury‘? Eur Heart J 2005; 26 : 99; author reply 99–100.
18. Kern KB, Hilwig RW, Berg RA et al. Postresuscitation left ventricular systolic and diastolic dysfunction. Treatment with dobutamine. Circulation 1997; 95 : 2610–2613.
19. Karimova A, Pinsky DJ. The endothelial response to oxygen deprivation: biology and clinical implications. Intensive Care Med 2001; 27 : 19–31.
20. Gando S, Nanzaki S, Morimoto Y et al. Out-of-hospital cardiac arrest increases soluble vascular endothelial adhesion molecules and neutrophil elastase associated with endothelial injury. Intensive Care Med 2000; 26 : 38–44.
21. Geppert A, Zorn G, Karth GD et al. Soluble selectins and the systemic inflammatory response syndrome after successful cardiopulmonary resuscitation. Crit Care Med 2000; 28 : 2360–2365.
22. Adrie C, Monchi M, Laurent I et al. Coagulopathy after successful cardiopulmonary resuscitation following cardiac arrest: implication of the protein C anticoagulant pathway. J Am Coll Cardiol 2005; 46 : 21–28.
23. Hékimian G, Baugnon T, Thuong M et al. Cortisol levels and adrenal reserve after successful cardiac arrest resuscitation. Shock 2004; 22 : 116–119.
24. Spaulding CM, Joly LM, Rosenberg A et al. Immediate coronary angiography in survivors of out-of-hospital cardiac arrest. N Engl J Med 1997; 336 : 1629–1633.
25. Lai CS, Hostler D, D‘Cruz BJ et al. Prevalence of troponin‑T elevation during out-of-hospital cardiac arrest. Am J Cardiol 2004; 93 : 754–756.
26. Müllner M, Hirschl MM, Herkner H et al. Creatine kinase-mb fraction and cardiac troponin T to diagnose acute myocardial infarction after cardiopulmonary resuscitation. J Am Coll Cardiol 1996; 28 : 1220–1225.
27. Nadkarni VM, Larkin GL, Peberdy MA et al. First documented rhythm and clinical outcome from in‑hospital cardiac arrest among children and adults. Jama 2006; 295 : 50–57.
28. Škulec R, Bělohlávek J, Dytrych V et al. Protokol pro použití terapeutické mírné hypotermie u nemocných po srdeční zástavě. Cor Vasa 2007; 49 : 61–65.
29. Hypothermia after Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med 2002; 346 : 549–556.
30. Bernard SA, Gray TW, Buist MD et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med 2002; 346 : 557–563.
31. Holzer M, Bernard SA, Hachimi-Idrissi S et al. Hypothermia for neuroprotection after cardiac arrest: systematic review and individual patient data meta‑analysis. Crit Care Med 2005; 33 : 414–418.
32. Lanier WL. Cerebral metabolic rate and hypothermia: their relationship with ischemic neurologic injury. J Neurosurg Anesthesiol 1995; 7 : 216–221.
33. Erecinska M, Thoresen M, Silver IA. Effects of hypothermia on energy metabolism in Mammalian central nervous system. J Cereb Blood Flow Metab 2003; 23 : 513–530.
34. Mori K, Maeda M, Miyazaki M et al. Effects of mild and moderate hypothermia on cerebral metabolism and glutamate in an experimental head injury. Acta Neurochir Suppl 1998; 71 : 222–224.
35. Graham SH, Shiraishi K, Panter SS et al. Changes in extracellular amino acid neurotransmitters produced by focal cerebral ischemia. Neurosci Lett 1990; 110 : 124–130.
36. Busto R, Globus MY, Dietrich WD et al. Effect of mild hypothermia on ischemia‑induced release of neurotransmitters and free fatty acids in rat brain. Stroke 1989; 20 : 904–910.
37. Kataoka K, Yanase H. Mild hypothermia – a revived countermeasure against ischemic neuronal damages. Neurosci Res 1998; 32 : 103–117.
38. Globus MY, Busto R, Lin B et al. Detection of free radical activity during transient global ischemia and recirculation: effects of intraischemic brain temperature modulation. J Neurochem 1995; 65 : 1250–1256.
39. Lei B, Tan X, Cai H et al. Effect of moderate hypothermia on lipid peroxidation in canine brain tissue after cardiac arrest and resuscitation. Stroke 1994; 25 : 147–152.
40. Edwards AD, Yue X, Squier MV et al. Specific inhibition of apoptosis after cerebral hypoxia-ischaemia by moderate post‑insult hypothermia. Biochem Biophys Res Commun 1995; 217 : 1193–1199.
41. Kumar K, Evans AT. Effect of hypothermia on microglial reaction in ischemic brain. Neuroreport 1997; 8 : 947–950.
42. Wang GJ, Deng HY, Maier CM et al. Mild hypothermia reduces ICAM‑1 expression, neutrophil infiltration and microglia/monocyte accumulation following experimental stroke. Neuroscience 2002; 114 : 1081–1090.
43. Deng H, Han HS, Cheng D et al. Mild hypothermia inhibits inflammation after experimental stroke and brain inflammation. Stroke 2003; 34 : 2495–2501.
44. Lee JE, Yoon YJ, Moseley ME et al. Reduction in levels of matrix metalloproteinases and increased expression of tissue inhibitor of metalloproteinase-2 in response to mild hypothermia therapy in experimental stroke. J Neurosurg 2005; 103 : 289–297.
Štítky
Dětská kardiologie Interní lékařství Kardiochirurgie KardiologieČlánek vyšel v časopise
Kardiologická revue – Interní medicína

2009 Číslo 1
Nejčtenější v tomto čísle
- Ischemicko-reperfuzní poškození po srdeční zástavě a protektivní účinky hypotermie
- Chirurgická léčba fibrilace síní
- Současný stav srdeční resynchronizační léčby u nemocných se srdečním selháním
- Galerie autorů