
Genotypově‑fenotypové korelace a stratifikace rizika náhlé srdeční smrti u familiární hypertrofické kardiomyopatie – kazuistika
Genotype ‑ phenotype correlation and risk stratification for sudden cardiac death in familial hypertrophic cardiomyopathy – a case study
Hypertrophic cardiomyopathy (HCM) is the most common genetic cardiac disease with a prevalence of 1 : 500. In familial cases the inheritance is autosomal dominant with non‑complete penetrance and variable expression; however, it can also be caused by de novo mutations. The heterogeneity of both its presentation and prognosis from case to case largely complicates clinical management, and therefore the disease often represents a dilemma for primary care clinicians as well as cardiologists. An estimation of sudden cardiac death risk is an integral part of clinical management and the stratification guidelines are continuously developing. We report on a case of familial HCM in two brothers with the same gene mutation, with very different clinical presentations and consequences.
Keywords:
familial hypertrophic cardiomyopathy – genetics – MYBPC3 – mutation – sudden cardiac death
Autoři:
A. Kilianová 1,2; M. Špinarová 1; L. Špinarová 1; I. Grochová 1; V. Feitová 2,3; J. Krejčí 1,2
Působiště autorů:
I. interní kardioangiologická klinika LF MU a FN u sv. Anny v Brně
1; Mezinárodní centrum klinického výzkumu, FN u sv. Anny v Brně
2; Klinika zobrazovacích metod LF MU a FN u sv. Anny v Brně
3
Vyšlo v časopise:
Kardiol Rev Int Med 2014, 16(6): 501-504
Kategorie:
Kardiologická revue
Souhrn
Hypertrofická kardiomyopatie (HCM) je nejčastější geneticky podmíněné srdeční onemocnění s prevalencí 1 : 500. Ve familiárních případech je dědičnost autozomálně dominantní s neúplnou penetrancí a různou expresivitou, může ovšem vznikat i de novo mutacemi. Nesmírná heterogenita projevů i prognózy HCM případ od případu značně komplikuje klinický management, a proto toto onemocnění stále často přináší mnoho otázek nejen pro lékaře prvního kontaktu, ale i pro kardiology. Stanovení rizika náhlé srdeční smrti u pacientů s HCM je integrální součástí klinického managementu a doporučení pro stratifikaci se neustále vyvíjí. V této kazuistice prezentujeme případ familiární HCM u dvou bratrů nesoucích stejnou mutaci s velmi rozdílným klinickým nálezem a průběhem.
Klíčová slova:
familiární hypertrofická kardiomyopatie – genetika – MYBPC3 – mutace – náhlá srdeční smrt
Úvod
Hypertrofická kardiomyopatie (HCM) je dle nových doporučení definována jako onemocnění charakterizované hypertrofií stěny levé komory srdeční (LKs), která není vysvětlitelná čistě hemodynamickými příčinami (jako např. hypertenzí či chlopenní vadou) [1]. Diagnostika je založena především na 2D echokardiografii, kde je kritériem tloušťka LKs ≥ 15 mm [2]. V posledních letech se stále více uplatňuje také magnetická rezonance, která umožňuje nejen přesnější morfologické rozlišení, ale také pomocí kontrastního vyšetření a metody pozdního sycení umožní znázornění myokardiální fibrózy jako negativního prognostického znaku [3]. Patofyziologie HCM je komplexní a zahrnuje obstrukci výtokového traktu levé komory (LVOT), diastolickou dysfunkci, mitrální regurgitaci, myokardiální ischemii a arytmie. Symptomy jsou velmi variabilní a většina pacientů zůstává celoživotně asymptomatických. Mezi typické příznaky patří bolesti na hrudi, námahová dušnost, synkopy, palpitace; někdy je prvním příznakem náhlá srdeční smrt (SCD). U malého procenta pacientů (5 %) může dojít k progresi onemocnění do tzv. end‑stage fáze s příznaky systolického srdečního selhání, kdy je jedinou radikální možností léčby srdeční transplantace. Terapeutický management u symptomatické hypertrofické kardiomyopatie zahrnuje medikamentózní léčbu (v ČR především betablokátory a kalciové blokátory), u obstrukčních forem s vysokým gradientem v LVOT rezistentních na farmakoterapii pak intervenční řešení nejčastěji alkoholovou septální ablací.
V histologickém obraze je patrna extenzivní hypertrofie kardiomyocytů a chaotické uspořádání hypertrofických svalových vláken, která se kříží a větví. Dále bývá zachycena intersticiální fibróza. Diferenciálně diagnosticky je nutno vyloučit především postižení srdce při hypertenzi, atletické srdce, amyloidózu srdce či střádavá onemocnění, jako je Fabryho nemoc či méně často Pompeho, Danonova či Gaucherova choroba [4,5].
Z molekulárně genetického hlediska jde o nejčastější geneticky podmíněné onemocnění srdce s celosvětovou prevalencí 1 : 500, což odpovídá v ČR asi 20 000 nemocných s HCM. Většina (60 %) mutací je autozomálně dominantních s různou expresivitou (tedy ne vždy se projeví ve výsledném fenotypu) a neúplnou penetrancí (ne vždy se fenotypově přenese na další generaci); tyto formy se označují jako familiární [1]. Mutace mohou vznikat ovšem i de novo v průběhu života. V současné době jsou známy stovky mutací na více než 80 genech, které jsou zodpovědné za vznik familiární HCM. V našem centru se nyní provádí v rámci panelu HCM analýza 46 genů metodou sekvenování nové generace.
Procentuálně nejčastěji jsou zastoupeny mutace v genech MYH7, MYBPC3 a TNNT2. Tyto geny kódují sarkomerické proteiny. Sarkomera je základní jednotkou srdeční kontrakce a skládá se z tenkých filament aktinu a tlustých filament myozinu, která se při kontrakci pohybují proti sobě.
Gen MYH7 kóduje beta těžký řetězec myozinu, který je součástí většího proteinu myozinu II. Myozin II se skládá ze dvou těžkých řetězců (produkovaných genem MYH7) a dvou párů regulujících lehkých řetězců (produkovaných několika jinými geny). Těžké řetězce mají dvě části: hlavu, která se označuje jako motorická doména a interaguje s tenkými filamenty aktinu, a tím je odpovědná za kontrakci srdeční buňky, a ocasní část, která interaguje s regulačními proteiny. Mutace v genu MYH7 jsou nejčastější příčinou familiární HCM s prevalencí až 35 %. Většina mutací genu MYH7 je způsobena záměnou jedné aminokyseliny, což vede ke vzniku defektního myozinu. Přesný mechanizmus, jakým tato mutace vede ke vzniku HCM, není znám [6].
Gen MYBPC3 kóduje myozin vazebný protein C (cardiac myosin binding protein C), který se podílí na stavbě sarkomer. Ve své aktivní fosforylované formě se váže na tlustá filamenta myozinu a zabraňuje jejich rozpadu. Mutace MYBPC3 genu jsou druhou nejčastější příčinou vzniku familiární HCM (30 % případů). Tyto mutace jsou zpravidla odpovědné za vznik abnormálně krátkého proteinu a vedou k hypertrofii srdečního svalu [7].
Gen TNNT2 kóduje srdeční troponin T, který je specifický pro srdeční sval. Srdeční troponin Tje jedním ze tří proteinů tvořící sarkomerický troponinový komplex, který společně s kalciem pomáhá regulovat srdeční kontrakci. Mutace v tomto genu se nachází asi u 5 % případů familiární HCM [8]. Mezi další poměrně časté geny patří TNNI3 (troponin I), TPM I (tropomyozin) a MYL3 (lehký řetězec myozinu 3) [8].
Familiární HCM je onemocnění typické svou heterogenitou, jak co se týče fenotypických projevů, tak i prognózy. Vzhledem k různé expresivitě a neúplné penetranci mohou zůstat někteří pacienti s prokázanými mutacemi v genotypu celoživotně bez vyjádřeného fenotypu, mohou však mít zvýšené riziko náhlé srdeční smrti. Proto je stratifikace rizika náhlé srdeční smrti jednou z nejdůležitějších součástí klinického managementu HCM. Nejaktuálnější studie u dospělých pacientů s HCM ukazují, že roční incidence úmrtí na kardiovaskulární příčinu je u této skupiny nemocných 1 – 2 % (tedy prakticky shodná jako u běžné populace), přičemž nejčastější příčinou je náhlá srdeční smrt (SCD), srdeční selhání a trombembolie [9]. Vysoce rizikových je asi 3 – 5 % nemocných s HCM [12]. Stanovení rizika by mělo zahrnovat osobní a rodinou anamnézu, 48hodinový EKG Holter, ECHO srdce a zátěžový test. Donedávna bylo hodnoceno několik charakteristik k určení rizika SCD a doporučení k implantaci kardioverteru ‑ defibrilátoru (ICD). Patřily zde záchyt nesetrvalé komorové tachykardie (NSVT), maximální tloušťka LKs ≥ 30 mm, rodinná anamnéza SCD, nejasná synkopa a abnormální tlaková reakce na zátěž [10]. Dle nejnovějších doporučení Evropské kardiologické společnosti se vypočítává individualizované riziko vzniku SCD do pěti let pomocí HCM Risk SCD kalkulátoru, který nezahrnuje abnormální tlakovou reakci na zátěž, ale zohledňuje např.výšku gradientu ve výtokovém traktu LK nebo velikost levé síně [1]. V současné době však neexistují žádné randomizované studie nebo statisticky validované prospektivní prediktivní modely, které by mohly být použity k rozhodnutí o implantaci ICD jako primární prevence SCD u pacientů s HCM, a doporučení se opírají o observační a retrospektivní studie, které stanovují souvislost mezi klinickými charakteristikami a prognózou.
Všechny tyto faktory značně komplikují klinický management, a proto HCM stále přináší mnoho dilemat jak pro lékaře prvního kontaktu, tak pro zkušené kardiology.
Kazuistika
V této práci prezentujeme případ dvou mladých mužů, bratrů, ve věku 28 a 36 let. Jejich rodinná anamnéza je zcela negativní. Mladšímu z bratrů, 28 let, byla již v dětství diagnostikována srdeční vada s hypertrofií stěn levé komory srdeční, genetické vyšetření v dětství však neodhalilo příčinu stavu. Z dalších onemocnění byla u pacienta zjištěna již v kojeneckém věku hypofunkce štítné žlázy, je na trvalé hormonální substituční terapii. Dále prodělal v dětství plané neštovice, absolvoval běžná očkování dle rozpisu, prodělal před lety tonsilektomii a operaci slepého střeva. V dlouhodobé medikaci užíval pouze levothyroxin 150 µg/ den. Je nekuřák. Na našem pracovišti byl poprvé vyšetřen ve věku 24 let, kdy byl plánovaně hospitalizován k provedení endomyokardiální biopsie (EMB) a pravostranné srdeční katetrizace v rámci došetření srdeční hypertrofie. Anamnesticky pacient udával občasné bušení srdce při větší námaze, jinak byl zcela bez potíží, bez bolesti na hrudi, bez dušnosti, bez jakéhokoli omezení v běžných aktivitách. Na vstupním EKG byly prokázány známky hypertrofie levé komory, obraz levého předního hemibloku a bloku pravého Tawarova raménka. Histologický obraz z EMB byl kompatibilní s diagnózou hypertrofické kardiomyopatie s roztroušenými hypertrofickými myocyty, místy se zmnožením fibrózní tkáně v intersticiu a nepravidelnostmi v paralelním uspořádání myocytů s jejich četnějším větvením. Speciálním barvením byly vyloučeny střádavé či infiltrativní choroby.
Za hospitalizace bylo též provedeno echokardiografické vyšetření (ECHO) srdce, kde byla potvrzena výrazná hypertrofie LKs, a to zejm. interventrikulárního septa s maximem ve střední části až 39 mm, systolicky téměř úplná okluze dutiny LKs ve střední části, EF 75 %, bez obstrukce LVOT. Dále byla v rámci stratifikace rizika SCD provedena ergometrie, která prokázala přiměřenou tlakovou reakci na zátěž, a Holterovo monitorování EKG, kde byly zachyceny pouze ojedinělé komorové extrasystoly (obr. 1).
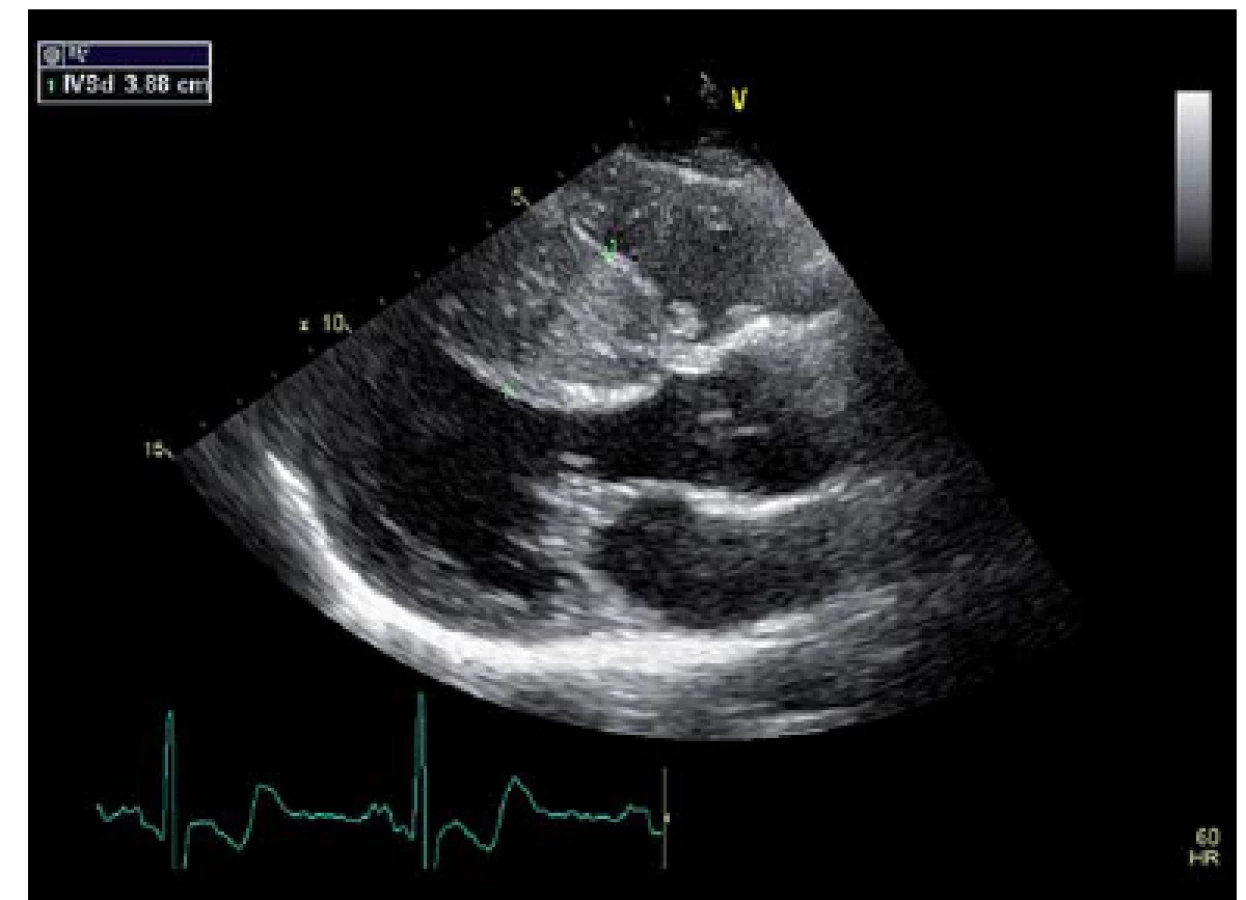
Pacient u nás zůstal ve sledování a byl pozván ke kontrolnímu ECHO vyšetření následující rok. Výsledek vyšetření byl stacionární od předešlé kontroly. Podobně kontrolní EKG Holter byl bez záchytu významných arytmií. Klinický stav pacienta se též výrazně nezměnil. Vzhledem k hereditárnímu charakteru tohoto onemocnění byl při této kontrole u pacienta proveden genealogický rozbor klinickým genetikem a následně molekulárně genetické vyšetření tehdy užívaného panelu pro familiární HCM zahrnující tři nejčastější geny MYH7, MYBPC3 a TNNT2 (vyšetření provedeno v roce 2010). U pacienta byla detekována kauzální mutace p.Thr.343MetfsX7c.1028delC genu MYBPC3, který kóduje myozin vazebný protein C.Tato mutace způsobuje vznik stop kodonu v exonu 12, a tedy zkrácení proteinového řetězce myozinového vazebného proteinu C.
Na základě této zjištěné kauzální mutace byl k vyšetření pozván i pacientův starší bratr, 32 let, který se dosud s ničím neléčil a neměl žádné kardiální obtíže. Genetická analýza prokázala i u něj stejnou mutaci p.Thr.343MetfsX7c.1028delC v genu MYBPC3. Echokardiografický nález byl však podstatně odlišný. Nález sice odpovídal obrazu HCM, ovšem tloušťka interventrikulárního septa byla „pouze“ 15 mm, EF 75 %, normální diastolická funkce. EKG bez patologického nálezu, EKG Holter a ergometrické vyšetření byly také negativní (obr. 2).

Oba bratři u nás zůstali ve sledování. Klinický stav i nález u staršího bratra s příznivým fenotypem je nadále stacionární, zůstává bez medikace či zvláštních opatření s výjimkou restrikce výkonnostních sportovních aktivit. U mladšího bratra s nepříznivým fenotypem přetrvává echokardiografický obraz významné hypertrofie interventrikulárnho septa s maximem 39 mm ve střední části, levá komora má typický banánovitý tvar. Na Holterově monitorování EKG byla zachycena intermitentní atrioventrikulární (AV) blokáda Mobitzova II. typu a na ergometrickém vyšetření byl zaznamenán pokles krevního tlaku na vrcholu zátěže, který se však při kontrolním vyšetření následující rok již neprojevil. V této době byla také provedena magnetická rezonance srdce, kde byla potvrzena asymetrická hypertrofie myokardu s maximem v oblasti septa a přední stěny. Dále byl nalezen postkontrastní enhancement charakteru degenerativní fibrózy myokardu patrný v septu a v bazálním segmentu volné stěny pravé komory, což lze považovat za negativní prognostický faktor [3]. V současné době je klinický stav pacienta stabilní, avšak objevuje se zvýšená únava a zadýchávání při větší námaze, funkční třída NYHA II (obr. 3).

U mladšího bratra s nepříznivým fenotypem bylo na základě zjištěných výsledků zvažováno zavedení ICD, jelikož bylo naplněno „velké“ kritérium hypertrofie dle starších evropských a nových amerických doporučení [10], nicméně další kritéria naplněna nebyla (až na přechodný jednorázově přítomný pokles TK při zátěži). Na základě nového doporučení Evropské kardiologické společnosti z roku 2014 [1] byl proveden výpočet rizika na podkladě ESC risk‑kalkulátoru (HCM Risk‑SCD), které vyšlo 2,23 %. Pacient byl zařazen do nízkorizikové skupiny, a k implantaci ICD tak indikován nebyl a zůstává nadále v našem sledování. U staršího bratra s méně výrazným fenotypem bylo kalkulované riziko 1,62 %.
Diskuze
Hypertrofická kardiomyopatie má prokázaný genetický podklad asi v 60 % případů, kdy se mutace buď dědí autozomálně dominantně jako familiární forma, nebo vznikají nezávisle de novo. I pro familiární formy je typická velká genová heterogenita, kdy jsou v současnosti známy již stovky mutací na cca 80 genech odpovědných za HCM. Diagnostiku značně komplikuje fakt, že mutace mají variabilní expresi a neúplnou penetranci. Navíc většina rodin má své vlastní tzv. „privátní“ mutace. Určení prognózy pacientů s pozitivním genotypem a negativním fenotypem je proto stále kontroverzním tématem. V současné době není v ČR rutinní molekulární diagnostika familiární hypertrofické kardiomyopatie běžně dostupná, přestože se jedná o nejčastější kardiální genetické onemocnění, a provádí se pouze v několika málo specializovaných laboratořích. V užívaném panelu HCM je nyní zahrnuto 46 genů, které jsou testovány metodou sekvenace nové generace. V naší kazuistice byla prováděna molekulárně genetická analýza v roce 2010, a proto byly zahrnuty pouze tři nejčastější geny – MYH7, MYBPC3 a TNNT2, které byly testovány Sangerovou metodou. U probanda i jeho staršího bratra byla zjištěna stejná kauzální mutace p.Thr.343MetfsX7c.1028delC genu MYBPC3, který kóduje myozin vazebný protein C. Tato mutace je spojována s lepší prognózou, pozdějším nástupem příznaků a menší hypertrofií než mutace v genu pro beta těžký řetězec myozinu (MYH7) [12]. Rozdíly ve fenotypu u těchto dvou bratrů mohou být způsobeny jak neúplnou expresivitou u bratra s příznivým fenotypem, tak případnou další mutací u bratra s hypertrofickým fenotypem, která nebyla při testování zachycena. V současné době se rozšířil panel molekulárně genetické diagnostiky HCM na 46 genů. Použití techniky sekvenování nové generace umožňuje zachytit více potenciálně kauzálních mutací, ale také zvyšuje počet zachycených polymorfizmů či mutací, které nejsou patognomonické pro tuto diagnózu.
Ačkoli někteří pacienti se zachycenými mutacemi nemají žádné fenotypické projevy, otázka zvýšeného rizika vzniku srdečního selhání a SCD u těchto pacientů je stále předmětem odborné diskuze. Většina pacientů s pozitivní mutací je heterozygotních, ale ve 3 – 5 % mohou pacienti nést dvě mutace stejného genu (homozygoti) nebo mohou nést více mutací na různých genech. Tyto případy jsou spojeny s významnějšími fenotypickými projevy zpravidla v mladším věku a větším rizikem komplikací včetně SCD [11]. Dříve hodně diskutovaný koncept tzv. maligních mutací (mutací spojených s vysokým výskytem SCD) nebyl potvrzen.
Oba bratři mají ve srovnání s běžnou populací mírně zvýšené riziko SCD (2,27 % a 1,62 %), které však nedosahuje hodnoty indikující implantaci ICD. Při stanovování rizika SCD je nutné mít na paměti, že s narůstajícím počtem rizikových faktorů se riziko SCD dramaticky zvyšuje – v nepřítomnosti rizikových faktorů je šestileté přežití 95%, naopak při třech rizikových faktorech pouhých 36 %, jak prokazují ve své práci Elliott et al [13]. Sledování nemocných by mělo u symptomatických pacientů probíhat každé tři měsíce, u asymptomatických jednou ročně, jeho součástí by vždy mělo být i zhodnocení rizika SCD.
Závěr
Tato kazuistika dokumentuje heterogenitu korelace mezi genotypem a fenotypem u familiární hypertrofické kardiomyopatie, kdy dva bratři nesoucí stejnou kauzální mutaci mají zcela odlišné projevy. Mladší z bratrů má hypertrofii srdce již od dětství, zatímco starší bratr má pouze hraniční tloušťku mezikomorového septa a je bez klinických obtíží. Dle kalkulovaného rizika náhlé srdeční smrti mají oba bratři pouze velmi mírně zvýšené riziko náhlé srdeční smrti ve srovnání s běžnou populací. Oba bratři referovaní v této kazuistice jsou prakticky asymptomatičtí s nízkým rizikem SCD, a proto zůstávají bez farmakologické i nefarmakologické intervence v našem klinickém sledování s pravidelným přehodnocování rizika SCD.
Brno Ph.D. Talent Scholarship Holder – Funded by the Brno City Municipality.
Supported by European Regional Development Fund – Project FNUSA ‑ ICRC (No. CZ.1.05/ 1.1.00/ 02.0123), European Social Fund and the State Budget of the Czech Republic.
Doručeno do redakce: 11. 10. 2014
Přijato po recenzi: 18. 11. 2014
MU Dr. Anna Kilianová
www.fnusa.cz
anna.kilianova@fnusa.cz
Zdroje
1. Elliott PM, Anastasakis A, Borger MA et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy, the task force for the diagnosis and management of hypertrophic cardiomyopathy of the european society of cardiology (ESC). Eur Heart J 2014; 35 : 2733 – 2779.
2. Maron BJ, Ommen SR, Semsarian C et al. Hypertrophic Cardiomyopathy Present and Future, With Translation Into Contemporary Cardiovascular Medicine. J Am Coll Cardiol 2014; 64 : 83 – 99. doi: 10.1016/ j.jacc.2014.05.003.
3. O’Hanlon R, Grasso A, Roughton M et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56 : 867 – 874.
4. Pelliccia A, Maron MS, Maron BJ. Assessment of left ventricular hypertrophy in a trained athlete: differential diagnosis of physiologic athlete’s heart from pathologic hypertrophy. Prog Cardiovasc Dis 2012; 54 : 387 – 396. doi: 10.1016/ j.pcad.2012.01.003.
5. Rapezzi C, Arbustini E, Caforio AL et al. Diagnostic work ‑ up in cardiomyopathies: bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2013; 34 : 1448 – 1458. doi: 10.1093/ eurheartj/ ehs397.
6. Genetics Home Reference: MYH7. [online] Available from: http:/ / ghr.nlm.nih.gov/ gene/MYH7.
7. Genetics Home Reference: MYBPC3. [online] Available from: http:/ / ghr.nlm.nih.gov/ gene/ MYBPC3.
8. Genetics Home Reference: TNNT2. [online] Available from: http:/ / ghr.nlm.nih.gov/ gene/ TNNT2.
9. Elliott PM, Gimeno JR, Thaman R. Historical trends in reported survival rates in patients with hypertrophic cardiomyopathy. Heart 2006; 92 : 785 – 791.
10. Gersh BJ, Maron BJ, Bonow RO et al. 2011 ACCF/ AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/ American Heart Association Task Force on Practice Guidelines. Circulation 2011; 124: e783 – e831. doi: 10.1161/ CIR.0b013e318223e2bd.
11. Genetic Testing Registry. Cardiomyopathy, familial hypertrophic type 4. [online] Available from: http:/ / www.ncbi.nlm.nih.gov/ gtr/ tests/ 510789/dne 10.11.2014.
12. Charron P, Dubourg O, Desnos M et al. Clinical features and prognostic implications of familial hypertrophic cardiomyopathy related to the cardiac myosin‑binding protein C gene. Circulation 1998; 97 : 2230 – 2236.
13. Elliott PM, Poloniecki J, Dickie S et al. Sudden Death in hypertrophic cardiomyopathy: identification of high risk patiens. J Am Coll Cardiol 2000; 36 : 2212 – 2218.
Štítky
Dětská kardiologie Interní lékařství Kardiochirurgie KardiologieČlánek vyšel v časopise
Kardiologická revue – Interní medicína

2014 Číslo 6
Nejčtenější v tomto čísle
- Choroby aorty – diagnostika, klasifikácia a princípy manažmentu
- Kombinace antikoagulační a antiagregační léčby u nemocných po infarktu myokardu indikovaných k antikoagulační léčbě – tzv. triple terapie
- Trojkombinace v léčbě hypertenze
- Kombinace ACE inhibitorů a sartanů – kdy je doporučena?